Synthesis of Phosphonate Derivatives of Benzisoselenazolones and Their Remarkable Antiureolytic Activity in Helicobacter pylori Cells
Marta Grabarek, Wojciech Tabor, Paweł Krzyżek, Julia Bąkowicz, Agnieszka Grabowiecka, Łukasz Berlicki, Artur Mucha

TL;DR
This paper describes the creation of new compounds that effectively block urease activity in Helicobacter pylori, a harmful bacteria, by combining specific chemical structures.
Contribution
The study introduces phosphonate derivatives of benzisoselenazolones with remarkable antiureolytic activity in Helicobacter pylori.
Findings
Esters of the compounds showed more substantial urease inactivation than expected.
Some compounds achieved IC50 values as low as 30–40 nM in Helicobacter pylori cells.
The compounds were synthesized through a multistep process involving aminolysis and hydrolysis.
Abstract
The attachment of a nickel-ion-complexing functionality to the structures of covalent inhibitors of ureases has been considered an effective method for enhancing binding to these pivotal virulence factors of various microbial pathogens. Following this approach, we envisioned a structural combination of 1,2-benzisoselenazol-3(2H)-one, a scaffold that produced the most significant antiureolytic effect achieved, with a phosphonic acid group intended to block the function of nickel ions in the catalytic mechanism. The multistep preparation of hybrid compounds involved aminolysis of 2-(chloroseleno)benzoyl chloride with the key diethyl aminophosphonate intermediates, followed by hydrolysis of the final phosphonate esters. Although not entirely consistent with the rationale of the design idea, the esters themselves, rather than the corresponding acids, demonstrated more substantial…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1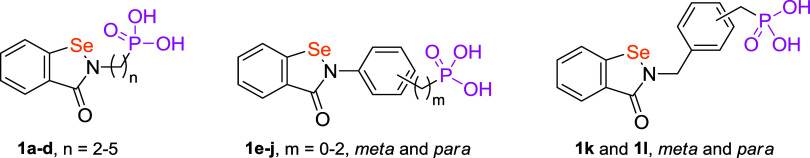 2
2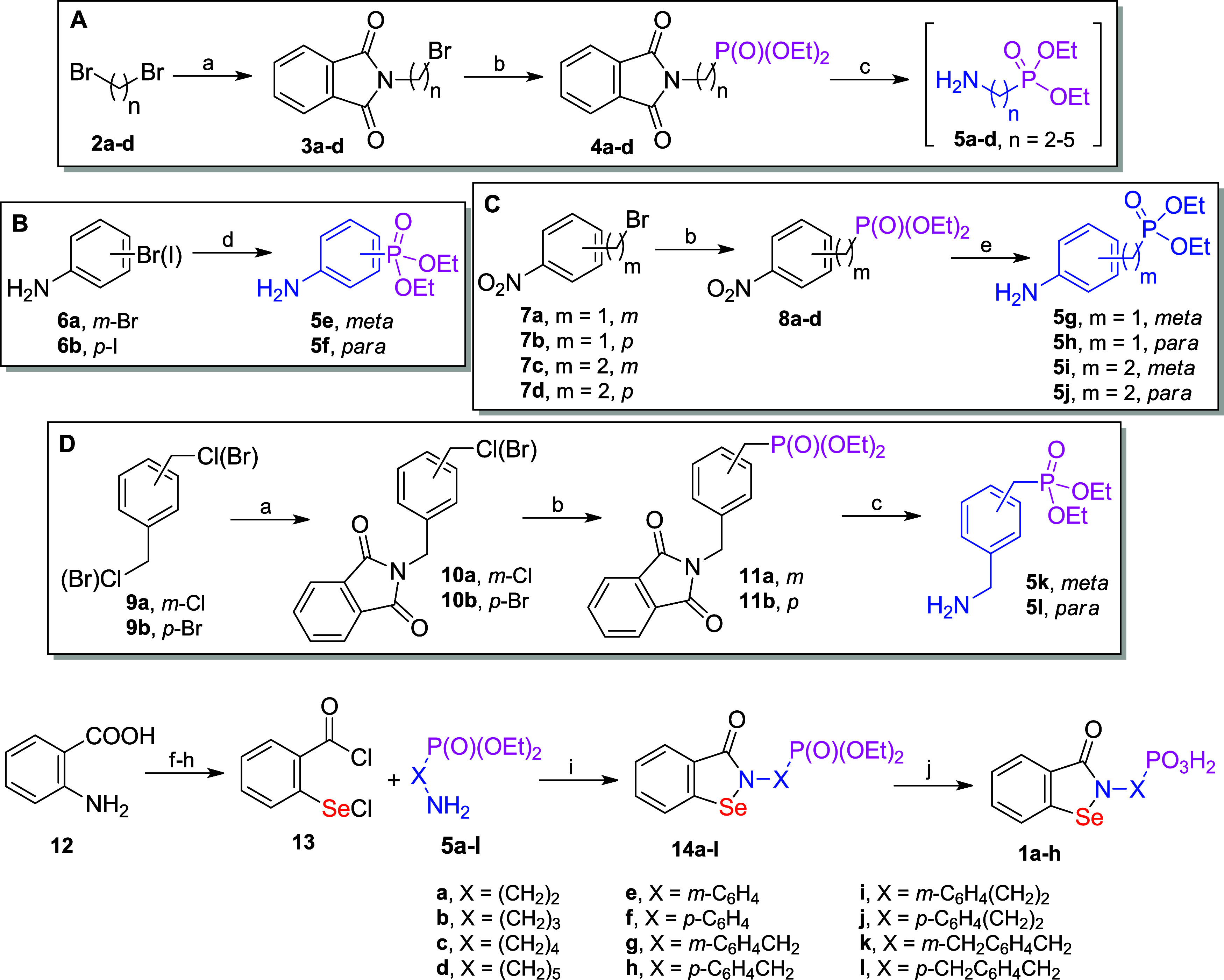 1
1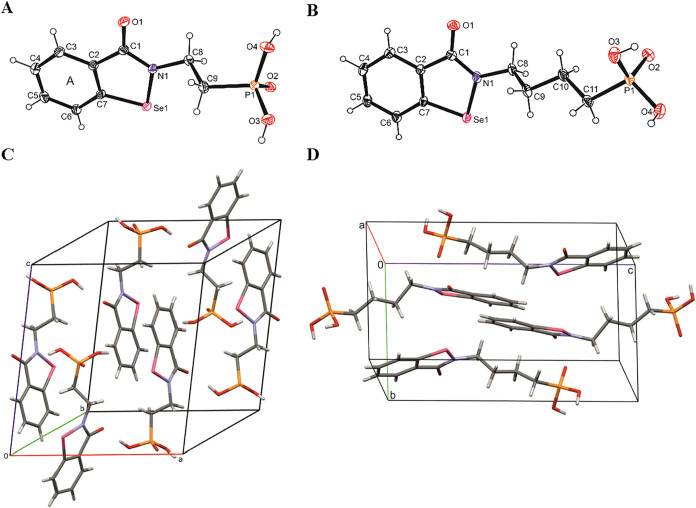 3
3 2
2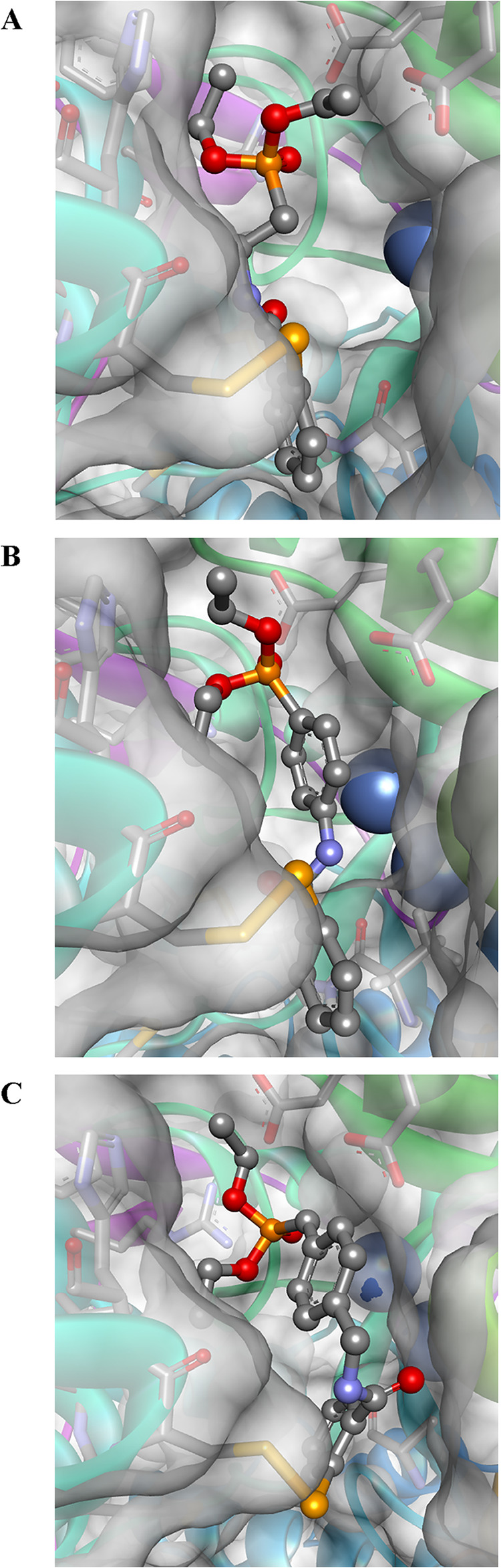 4
4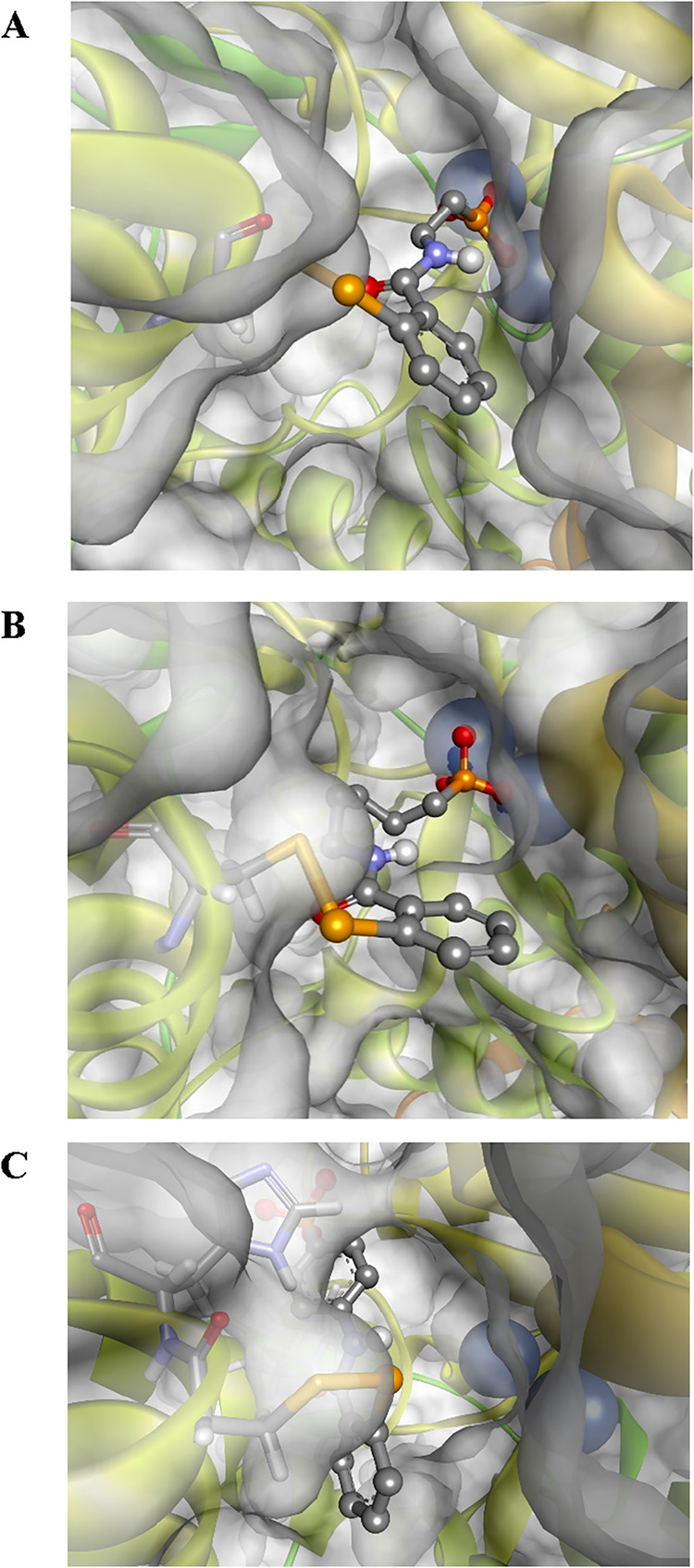 5
5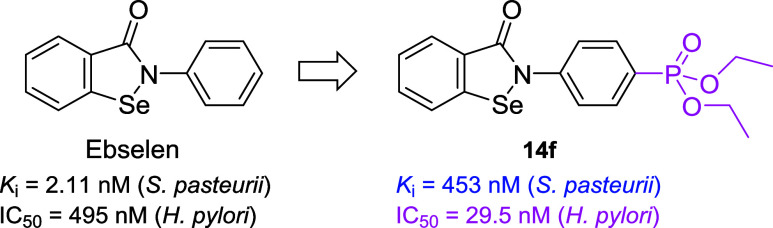 6
6| retained
enzyme activity [%], DTT added 0.5 h after the reaction initialization | |||
|---|---|---|---|
| inhibitor ID | retained enzyme activity [%], DTT added before the reaction initialization | no enzyme–inhibitor preincubation | 2 h enzyme–inhibitor preincubation |
|
| 100 | 27.3 ± 0.7 | 0 |
|
| 100 | 31.9 ± 3.0 | 0 |
|
| 100 | 100 | 36.5 ± 1.5 |
|
| 100 | 76.3 ± 3.5 | 59.6 ± 3.9 |
|
| 80.4 ± 2.7 | 53.8 ± 0.4 | 0 |
|
| 86.2 ± 3.1 | 67.7 ± 7.2 | 0 |
|
| 53.1 ± 0.8 | 51.8 ± 4.1 | 0 |
|
| 56.7 ± 5.1 | 53.5 ± 1.9 | 13.5 ± 0.6 |
| compound ID | MIC |
|---|---|
|
| 80 μM (33.0 μg/mL) |
|
| 20 μM (8.24 μg/mL) |
|
| 40 μM (17.0 μg/mL) |
|
| 80 μM (34.1 μg/mL) |
|
| 80 μM (29.6 μg/mL) |
|
| 40 μM (17.6 μg/mL) |
| amoxicillin | 0.015 μg/mL |
| MIC [μg/mL] | ||||||
|---|---|---|---|---|---|---|
| compound ID | alone | combination | fold change | FIC | FICI | interpretation |
|
| 8.24 | 4.12 | ×2↓ | 0.5 | 0.75 | additive |
| amoxicillin | 0.015 | 0.038 | ×4↓ | 0.25 | ||
|
| 8.24 | 2.06 | ×4↓ | 0.25 | ||
| amoxicillin | 0.015 | 0.075 | ×2↓ | 0.5 | ||
| IC50/IC50 95CI [μM] | ||
|---|---|---|
| compound ID | Balb/3T3-L1 | HEK-293 |
|
| 65.6/51.0–84.0 | 72.5/19.4–195 |
|
| >100 | 39.8/24.2–51.2 |
|
| 76.9/52.3–127 | >100 |
|
| 43.7/35.3–54.4 | 62.5/26.7–177 |
|
| 44.8/33.3–61.8 | 63.0/42.0–103 |
|
| 64.6/54.3–76.9 | 42.4/31.4–59.1 |
|
| 48.4/39.3–60.4 | 61.8/42.5–95.7 |
| cisplatin | 3.30/2.69–4.11 | 0.142/0.022–0.319 |
- —Narodowe Centrum Nauki10.13039/501100004281
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSynthesis and Reactivity of Sulfur-Containing Compounds · Organophosphorus compounds synthesis · Synthesis and biological activity
Introduction
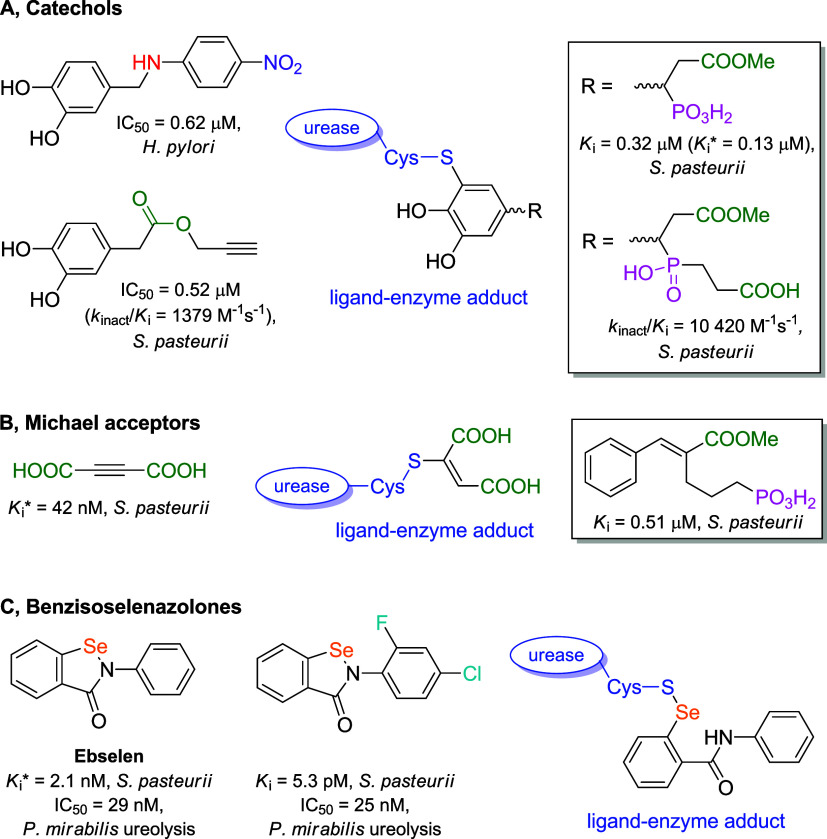
Covalent inhibition based on the specific reactivity of functionalized low-molecular-weight organic compounds has recently emerged as a promising approach to effectively control the activity of bacterial ureases. Urease,? a landmark enzyme in advances in biochemistry and medicine, ?,? is a hydrolase that catalyzes amide bond cleavage in urea to form ammonia and carbamate; then, carbamate hydrolyzes spontaneously. The general process of decomposing urea to ammonia and carbon dioxide that occurs in plants, fungi, and bacteria is an important element of the global circulation of nitrogen.? The anthropogenic aspect of this system mainly reflects the issue of nitrogen fertilizers and the limitation of their efficacy due to hydrolysis by macrobiotic ureases in the soil. Nitrogen losses require extensive use of urea, while the overproduction of ammonia as a direct result of uncontrolled ureolysis in the environment leads to water and air pollution and water eutrophication. ?,? With respect to human health, the importance of bacterial ureolysis lies in the medical potential of its control as a virulence factor of persistent pathogenic microorganisms, such as Proteus mirabilis or Helicobacter pylori, which infect the urinary and gastrointestinal tracts, respectively.? Products of ureolytic activity are involved in the formation of urinary kidney stones? or allow bacteria to survive in the acidic environment during colonization and induce chronic gastritis and peptic and duodenal ulceration, with the risk of gastric cancers.? Inhibition of urease activity reduces the virulence, colonization abilities, and viability of pathogens and increases their susceptibility to first-line antibiotics. ?,?,?
In light of the growing interest in covalent drugs over the last two decades, ?−? ? ? particularly those reacting with the cysteine residue,? urease has been viewed as an excellent antibacterial target, susceptible to inactivation based on selective reactivity. Three categories of compounds, namely, catechols, Michael acceptors, and organoselenium derivatives, have been successfully utilized for this purpose. ?−? ? Research on catechols as potential inhibitors of bacterial ureases was inspired by previous findings related to the inhibition of ureolysis in soil? and in plants (jack bean)? that involved natural polyphenolic structures. Accordingly, catechins, secondary antioxidant flavonoid metabolites found in green tea extracts, were shown to inactivate H. pylori urease (e.g., (−)-epigallocatechin gallate, IC_50_ = 2.2 μM).? Subsequently, various activities (typical IC_50_ values at moderate micromolar concentrations) were measured for other flavonoids isolated from natural sources and/or synthetically modified. ?−? ? Parallel SAR studies of a series of synthetic isoflavone-based pyrogallol and catechol derivatives identified the 1,2-diphenylethane core and two ortho hydroxyl groups as essential structural/functional elements for their activity against H. pylori urease. ?,? Further optimization of this scaffold yielded derivatives with increased affinity, such as 4-((4-nitrophenylamino)methyl)benzene-1,2-diol (IC_50_ = 0.62 μM for cell-free H. pylori urease; IC_50_ = 1.92 μM for ureolysis in intact cells; Figure, panel A). ?,? Pagoni et al. identified propargyl 3,4-dihydroxyphenylacetate, an active, covalent, and irreversible inhibitor of Sporosarcina pasteurii urease (IC_50_ = 0.52 μM; Figure, panel A), among 40 amide and ester derivatives of 3,4-dihydroxyphenylacetic acid, caffeic acid, ferulic acid, and gallic acid.? In the most recent studies, to enhance noncovalent interactions with catalytic nickel ions and surrounding basic amino acid residues, catechol reactivity was combined with the presence of phosphonic/phosphinic and carboxylate moieties. ?,? The products were shown to be effective ligands and demonstrated complex binding kinetics. For some of these, such as 1-(3,4-dihydroxyphenyl)-2-methoxycarbonylethylphosphonic acid, the inactivation process was slow but reversible (K i = 0.32 μM, dissociation constant of the initial enzyme–inhibitor complex; and K i* = 0.13 μM, overall dissociation constant; Figure, panel A, frame), while others, like 1-(3,4-dihydroxyphenyl)-2-methoxycarbonylethyl(2-carboxyethyl)phosphinic acid, were typically irreversible (k inact/K I = 10,420 s^–1^ M^–1^; Figure, panel A, frame).? The specific mechanism of ureolysis inhibition contributed to the safety and nontoxicity of catechol-based phosphonic and phosphinic acids against mammalian cells.? Ciurli et al. provided fundamental kinetic, computational, and structural data supporting the inactivation mechanism of ureases by catechols. ?,? The authors proposed a multistep radical process leading to the substitution of the aromatic ring with the reactive thiol group of the cysteine residue to create the covalent product (Figure, panel A). The key cysteine residue, for instance, Cys322 in S. pasteurii and Cys321 in H. pylori (numbering of the α subunit of (αβγ)3 or ((αβ)3)4 quaternary arrangement, ?,? respectively), is located within the loop of the movable helix–loop–helix flap that changes its conformation to accommodate the substrate for hydrolysis.? Covalent modifications hinder the access of urea to the active site and disrupt the adoption of the closed conformation necessary for catalysis.?
Background of the study. Structures of representative examples of covalent inhibitors of bacterial ureases, their activities against isolated enzymes (indicated by the systematic name of the microorganism) or ureolysis performed by pathogenic live cells, and proposed binding modes (ligand-enzyme adducts): catechol derivatives (panel A), Michael acceptors (panel B), and 1,2-benzisoselenazol-3(2H)-ones (panel C). Modifications containing a phosphorus atom are shown within the frames (for benzisoselenazol-3(2H)-ones, the focus of the current work).
The second group of covalent inhibitors of ureases consists of α,β-unsaturated compounds that undergo nucleophilic addition with the sulfhydryl group of the key cysteine residue (Michael addition). This activity was first demonstrated to retard the hydrolysis of urea in soil with p-benzoquinone.? Regarding bacterial enzymes, differently substituted benzo- and naphthoquinones noncompetitively and irreversibly inhibited S. pasteurii urease and ureolysis in the whole cells of H. pylori, Klebsiella oxytoca, and P. mirabilis.? p-Benzoquinone was also confirmed as the most active among 70 commercially available compounds screened against the ureolytic bacterium Klebsiella pneumoniae.? However, the application of quinones is not beneficial, as it is associated with low selectivity issues that result in cytotoxicity.? Macegoniuk et al. identified potent inactivators of S. pasteurii urease among a series of 40 α,β-unsaturated carbonyl and carboxyl compounds.? Low nanomolar values of steady-state inhibition constants (slow-binding mechanism of action, K i* ∼ 10 nM) were calculated for methyl and ethyl esters of acetylenedicarboxylic acid. The acid itself was somewhat less potent (K i* = 42 nM; Figure, panel B) but highly specific, as it demonstrated low chemical reactivity with alternative thiols and no cytotoxicity with mammalian cells. Its modeled binding mode revealed simultaneous addition with the Cys322 sulfhydryl group and coordination of a carboxylate with nickel ions of the active site. To further explore this cooperativeness, we combined the reactive α,β-unsaturated structure of cinnamate with phosphonic/phosphinic functionalities that were expected to enhance the complexation abilities of the products. ?,? As a result, a slow-binding submicromolar inhibitor of S. pasteurii urease (K i = 0.51 μM; Figure, panel B, frame) was identified; it also affected the whole cell-mediated ureolysis of P. mirabilis at the micromolar level (IC_50_ = 11 μM, with a 2 h preincubation time).
2-Substituted 1,2-benzisoselenazol-3(2H)-ones, derivatives of Ebselenan antioxidant, anti-inflammatory, cytoprotective organoselenium compound that reacts with thiols, ?,? represent the most recently developed and powerful group of covalent inhibitors of bacterial ureases. Ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one) was initially characterized as a potent competitive binder of the urease of S. pasteurii (K i* = 2.1 nM; Figure, panel C).? Substitution of the 2-phenyl ring, particularly dihalogenation, yielded derivatives that demonstrated an impressive three orders of magnitude improvement in affinity compared to the lead structure (e.g., 2-(4-chloro-2-fluorophenyl)-1,2-benzisoselenazol-3(2H)-one, K i = 5.3 pM; Figure, panel C).? The mentioned compounds effectively controlled ureolysis in live pathogenic P. mirabilis cells at remarkably low concentrations (IC_50_ = 29 nM for Ebselen and IC_50_ = 25 nM for 4-chloro-2-fluoro modification). However, this effect was not observed in H. pylori cells. Recently, we published data on halogenated 2-benzyl benzisoselenazolones.? These compounds altered the ureolytic activity of live H. pylori at IC_50_ < 100 nM, as reported for the first time. Other modified organoselenium derivatives of this framework, namely, 2-aryloyl benzisoselenazolones and benzoselenazolyl benzoates, were currently targeted against ureolysis performed by H. pylori.? The best newly synthesized covalent inhibitor exhibited an IC_50_ = 0.14 μM against the free enzyme and MIC = 8 μg/mL with pathogenic cells. The commonly accepted mechanism of action of Ebselen and its derivatives on proteins involves the cleavage of the Se–N bond in the benzisoselenazolone ring by a nucleophilic thiol/thiolate of the cysteine residue, forming a new S–Se bond (Figure, panel C).? The reaction is rapid and efficient, selective against other nucleophiles, and can be reversed by adding low-molecular-weight thiols. The formation of the enzyme–inhibitor complex is typically facilitated by extensive hydrophobic interactions with the protein surface.? This mode of action was also proposed for the inhibition of urease by Ebselen and was supported by molecular modeling studies.? However, the initial complex may undergo aging due to its S_N_Ar-like nucleophilic hydrolysis, releasing salicylanilide and the selenated enzyme as recently observed for the inactivation of M^pro^, the main protease of SARS-CoV-2,? and then S. pasteurii urease.?
In the current work, we envisaged using the 1,2-benzisoselenazol-3(2H)-one structure for synthesizing hybrid compounds modified with a phosphonic acid functionality at the terminus of 2-substituents. To classify the results in the context of previous data, we first studied these compounds with the model bacterial urease of S. pasteurii; however, efficient inhibition of ureolysis in whole H. pylori cells remained the most challenging goal.
Results and Discussion
Compounds
Among the three discussed groups of covalent inhibitors of bacterial ureases (catechols, Michael acceptors, and organoselenium derivatives), benzisoselenazolones, although not antibiotics as such, appear to exhibit the most significant potential for developing effective antimicrobial therapies through a repurposing approach.? The anti-inflammatory, antioxidant, and cytoprotective activities of Ebselen, the prototypical representative, are well characterized; ?,? the same holds for its toxicity and pharmacological properties.? On the other hand, the fundamental activity of this compound in bacteria is specific and involves inhibition of thioredoxin reductase, which alters the reduction of disulfides in multiple substrates, influences cell redox status, impacts the biosynthesis of DNA and cellular proteins, and induces oxidative stress.? Beneficially, these effects can be enhanced by inactivating bacterial virulence factors, such as urease.? Employing these principles, we proposed functionalizing the advantageous 1,2-benzisoselenazol-3(2H)-one structure with a phosphonic acid functionality (Figure). This group was intended to be located at the 2-substituent to facilitate the formation of additional ligand–protein interactions, particularly the complexation of catalytic nickel ions. Noncovalent blocking of the catalytic function of the metal ions by phosphorus-based acid has been proven to be one of the most effective approaches to inhibit microbial ureases. ?−? ? As mentioned above, the corresponding functionalization of catechols and Michael acceptors resulted in covalent enzyme inactivators with improved characteristics. To perform these modifications, a series of 2-substituents was utilized: alkyl, aryl, or arylalkyl (compounds 1a–l; Figure); their length and regioisomerism varied widely to bridge the gap between the target sites in the enzyme structure: the cysteine residue in the movable flap and the central nickel ions.
Designed structures: phosphonic acid-modified 2-alkyl, aryl, or arylalkyl 1,2-benzisoselenazol-3(2H)-ones that contain a selenium heterocycle reactive with cysteine thiolate located within the movable flap at the entrance to the active site of urease, along with a phosphonic group intended for forming noncovalent ligand–protein interactions, particularly complexation with nickel ions.
Synthesis and Structure
The synthesis of the target compounds was based on the aminolysis of 2-(chloroseleno)benzoyl chloride with aminophosphonates 5a–l of the designed structures (Scheme). To achieve these key aminophosphonate scaffolds, the phosphonate group was introduced to suitable alkyl or aryl halide substrates using classical approaches, namely, the Michaelis–Arbuzov reaction? or palladium-catalyzed cross coupling,? respectively. Amino functionalization, in turn, involved Gabriel synthesis (for the alkyl amino group)? or reduction of the nitro group (for the aryl amino group),? as outlined in panels A–D. Specifically, diethyl ω-aminoalkylphosphonates 5a–d were obtained from α,ω-dibromoalkanes 2a–d in a three-step synthetic route via 2-(ω-bromoalkyl)phthalimides 3a–d and the corresponding diethyl ω-phthalimidoalkylphosphonates 4a–d through two consecutive substitutions with potassium phthalimide and triethyl phosphite, followed by hydrazinolysis of imides 4a–d (Scheme, panel A). The Arbuzov reaction step was aided by microwave induction,? which allowed for a short heating time and resulted in excellent yields. As chromatography purification of 5a–d did not enhance the quality of the final materials (due to partial ester hydrolysis), the crude products after hydrazinolysis were used in the subsequent aminolysis of chloride 13.
Multistep Synthesis of Diethyl Phosphonate and Phosphonic Acid Derivatives of 2-Substituted Benzisoselenazolones, Including the Preparation of Key Intermediate Aminophosphonates (Panels A–D, Frames)
To obtain diethyl meta- and para-aminophenylphosphonates (5e and 5f, Scheme, panel B), two alternative conditions for palladium-catalyzed coupling with a phosphorus component were compared. The Hirao reaction, starting with m-bromoaniline (6a) and diethyl phosphite,? proved to be more effective than the coupling of iodoaniline (6b) and triethyl phosphite under aqueous conditions.? Diethyl meta- and para-aminobenzyl- and aminophenetylphosphonates (5g–j) were synthesized from nitrophenylalkyl bromides 7a–d in the Arbuzov reaction, followed by the reduction of diethyl nitrophenylalkylphosphonates 8a–d with tin(II) chloride (Scheme, panel C). Finally, diethyl meta- and para-(aminomethyl)benzylphosphonates (5k and 5l) were obtained from xylylene halides (9a and 9b) via (phthalimidomethyl)benzyl halides (10a and 10b) and (phthalimidomethyl)benzylphosphonates (11a and 11b), combining the analogous transformations described above for diethyl alkylphosphonates 5a–d (Scheme, panel D). Several aminophosphonate compounds mentioned have been reported in the literature. ?−? ? ? ? ? ? Nevertheless, the preparation details, along with a comprehensive characterization of all structures (except aminoesters 5a–d, used as crude) and their precursors, both known and previously unreported, are provided in the Supporting Information.
The resulting diethyl aminophosphonates 5a–l were subjected to reactions with 2-(chloroseleno)benzoyl chloride (13), the latter obtained from anthranilic acid (12), carried out according to the standard procedure. ?,? The entire series of final diethyl phosphonate derivatives of 2-substituted benzisoselenazolones 14a–l was separated with good yields after workup and chromatographic purification. Subsequently, several conditions for acidic hydrolysis or dealkylation (TMSBr) of phosphonate esters were tested to ensure practical yields of free phosphonic acids. A mixture of acetic acid and concentrated hydrochloric acid, heated briefly in a microwave oven at 140 °C, was found to be the most convenient approach. This method avoided extensive isoselenazolone ring opening, the main unwanted acid-mediated side reaction. After chromatographic purification, some target products yielded well-defined crystals (Figure). Compound 1a crystallized in the triclinic space group P1̅ with three symmetrically independent molecules, A, B, and C, in the asymmetric unit (only one of them, molecule A, is presented in Figure, panel A), while compound 1c (Figure, panel B) crystallized in the monoclinic space group P2_1_/n.
Molecular structure of phosphonic acids 1a and 1c with the atom-numbering scheme (panels A and B). Displacement ellipsoids are drawn at the 25% probability level, and H atoms are represented as small spheres with arbitrary radii. The arrangement of the molecules in the unit cells was prepared using the Mercury program (panels C and D). Crystallographic data for the structures have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 2323717 and CCDC 2323716.
Regarding the supramolecular arrangement, the structures of compounds 1a and 1c revealed somewhat different organizations and modes of interaction (Figure, panels C and D, respectively). In fact, in both cases, all three oxygen atoms of the phosphonic acid group were involved in hydrogen contacts; two of them included the adjacent acid functionality(ies), while the third one pointed toward the oxo substituent of the neighboring isoselenazole ring. However, for the structure of 1a, the contact between the phosphonic acid groups was bidentate and formed a dimeric arrangement, while for the structure of 1c, the interactions involved the groups located above and below, linking a stack of molecules perpendicularly to their planar surface. The hydrophobic benzisoselenazolone fragments of 1a molecules, alternately inverted in opposite directions, formed π-stacking piles. In the layers of 1c, nonpolar contacts involved an interchangeable superposition of the alkyl on the aryl part of the molecules.
We decided against performing hydrolysis for all members of the compound set. When tested for inhibition of ureolysis, somewhat surprisingly and contrary to design principles, the acids appeared significantly less active than their esterified counterparts, in several instances even by an order of magnitude (vide infra). Therefore, we deemed it unnecessary to remove the ethyl groups from the last four compounds, 14i–l, of the series. Instead, we explored the potential of selective hydrolysis. A single phosphonate alkyl residue could be cleaved under basic conditions (Scheme). Indeed, it performed satisfactorily, and two exemplified benzisoselenazolones were obtained and characterized: monoethyl ethylphosphonate 15a, the analogue of 14a and 1a, and monoethyl pentylphosphonate 15b, the analogue of 14d and 1d.
Selective Hydrolysis of Chosen Diethyl Phosphonates to Obtain Monoethyl Esters of Phosphonic Acids
Antiureolytic Activity
The first group of compounds studied, 2-alkyl derivatives of 1,2-benzisoselenazol-3(2H)-one containing diethyl phosphonate, phosphonic acid, or monoethyl phosphonate functionality at the ω position (14a–d, 1a–d, 15a, and 15b, respectively), exhibited a quite unexpected structure–activity relationship. Phosphonate diesters 14a–d were found to act as excellent inhibitors of purified urease from S. pasteurii (K i = 12.8–71.6 nM) and of urea decomposition in live cells of H. pylori (IC_50_ = 130–299 nM; Table). The values of the kinetic parameters varied within a narrow range. For S. pasteurii urease, these changes were irregular with the length of the alkyl chain linker; nevertheless, the most extended compound (14d) was found to act as the most potent inhibitor. For H. pylori cells, the effectiveness of the ureolysis control decreased from the shortest homologue (14a) to the longest homologue (14d). To contextualize these data within the literature, a much more striking level of inhibition of S. pasteurii urease was previously reported with Ebselen? and its derivatives,? although the alteration of ureolytic activity in H. pylori described in this work reached nearly top levels.?
1: Inhibitory Activity of Synthesized Compounds against Urease of S. pasteurii and Ureolysis of Live Cells of H. pylori Tx30a (the Most Significant Data Are Indicated in Bold)
Surprisingly, phosphonic acids 1a–d did not follow the remarkable characteristics of their parent diethyl esters 14a–d. These acids were several times, up to an order of magnitude, weaker inhibitors in the two systems studied (K i = 108–426 nM for S. pasteurii urease and IC_50_ = 867–2106 nM for H. pylori ureolysis, Table). In both assays, the most notable kinetic parameters were acquired for the 2-(5-phosphono)pentyl analogue 1d. Although objectively significant, the results suggested that the covalent binding of benzisoselenazolones was not facilitated by cooperative interactions of the phosphonic acid moiety. Partial hydrolysis of selected diethyl esters was also found to be ineffective with regard to potency. Monoethyl esters of phosphonic acids (15a and 15b) demonstrated a further decrease in antiureolytic activity compared to the corresponding diethyl esters (14a and 14d) and acids (1a and 1d), as evidenced by micromolar values of measured kinetic constants (Table).
Studies of meta- and para-2-phenyl benzisoselenazolones, diethyl phosphonates 14e and 14f, and phosphonic acids 1e and 1f revealed a structure–activity relationship distinct from that discussed for 2-alkyl derivatives. The four compounds were practically equipotent toward S. pasteurii urease (K i = 286–453 nM; Table), indicating good but decreased potency compared to compounds 14a–d. For antiureolytic activity in H. pylori, the differentiation of phosphonate esters and phosphonic acid, by an order of magnitude of the IC_50_ value, became evident again. Furthermore, the control of urea decomposition by esters 14e and 14f in pathogen cells reached an unprecedented level, with IC_50_ at a nanomolar concentration: IC_50_ = 40.4 ± 0.62 nM for meta analogue 14e and IC_50_ = 29.5 ± 0.34 nM for its para regioisomer 14f. To the best of our knowledge, these IC_50_ values were lower than any other data in the literature reporting inhibition of ureolysis in native H. pylori.
The final structural combinations involved the installation of the diethyl phosphonate functionality at the termini of the 2-benzisoselenazolone substituents, which comprised alkyl and aryl fragments (meta and para regioisomers), such as benzyl (14g and 14h), phenethyl (14i and 14j), and xylyl (14k and 14l). Addition of one flexible methylene linker prompted a visible improvement in inhibiting the activity of S. pasteurii urease by benzylphosphonates 14g and 14h compared to sterically rigid phenylphosphonates 14e and 14f (K i = 7.77 ± 2.8 nM versus 286 ± 34 nM for meta homologues and K i = 105 ± 8.5 nM versus 453 ± 15 nm for para homologues), while the potency of the products of the hydrolysis, phosphonic acids 1g and 1h, remained practically the same as that measured for 1e and 1f. In H. pylori cells, esters 14g and 14h exhibited excellent potency, characterized by IC50 values that are only twice that of 14f. As the ester hydrolysis of compounds 14g and 14h did not enhance the activity of the resulting phosphonic acids 1g and 1h, we discarded this step for the remaining four structures and focused solely on phosphonates.
Further structural extension and increased flexibility resulted in the optimized potency of diethyl esters 14i–l. These compounds demonstrated a high affinity for S. pasteurii, as indicated by low nanomolar K i values (below 68 nM), with phosphonate 14k being the most potent inhibitor identified in this study (K i = 5.06 ± 0.42 nM). At the same time, three of them (14j–l) maintained the top antiureolytic effect in H. pylori cells (IC_50_ = 32.2–33.5 nM), comparable to that found for the 2-aryl derivative 14f. In both systems studied, xylyl derivatives 14k and 14l, which possess two benzyl-type connections at the nitrogen atom of benzisoselenazolone and the phosphonate functionality, acted more efficiently than their structural isomers that comprised an ethylene linker (14i and 14j). The reasons might be differences in the reactivity of the isoselenazolone system (N-benzyl versus N-aryl-substituted) or a potential to form noncovalent interactions with the enzymes (a lower steric rigidity of the xylyl system).
Finally, a certain regularity in the structure–activity relationship of phosphonate esters that include a phenyl fragment in their 2-substituent (14e–l) could be observed. Within each pair of compounds, the meta regioisomer exhibited a higher affinity in inhibiting S. pasteurii urease than its para counterpart (14e vs 14f, 14g vs 14h, 14i vs 14j, and 14k vs 14l). Interestingly, the influence on ureolysis in H. pylori cells showed a consistently opposite behavior pattern. In most cases, the changes are subtle, and we would rather avoid further generalization.
Binding Kinetics
Despite the structural and activity differences between diethyl phosphonates 14a–l and phosphonic acids 1a–h, all of these compounds demonstrated comparable binding kinetics to the urease of S. pasteurii (see the Supporting Information, Figures S1 and S2). The progress curves of the inhibited reactions without enzyme–inhibitor preincubation were nonlinear, and the initial and steady-state velocities were inversely proportional to the concentration of benzisoselenazolones. The inhibitors were determined to be slow-binding and operated according to a two-step binding mechanism: a relatively rapid formation of enzyme-ligand complexes was followed by a limiting step of slow conformational changes, plausibly necessary for the formation of the S–Se bond. The binding mechanism of selected derivatives (esters 14e–h and acids 1e–h) was further studied through experiments that utilized dithiothreitol (DTT) as an agent that protected/reinstated the urease activity in the presence of inhibitors that bind to the thiol group of the cysteine in the active site. The inhibitors studied in this experiment were used at a saturating concentration of 5 μM, and the DTT concentration in all experiments was twice as high. First, DTT was added simultaneously to the inhibitor to assess its ability to disrupt the initial stage of EI complex formation (enzyme protection). No inhibitory activity was observed for compounds 14e, 1e, 14f, and 1f (Table). However, for both diethyl benzylphosphonates 14g and 14h, as well as phosphonic acids 1g and 1h, the velocity of ureolysis was only partially maintained (53.1–86.2%). It is noteworthy that benzylphosphonate esters 14g and 14h, in particular, were much more active inhibitors of bacterial ureases than their homologues 14e and 14f. In subsequent studies, DTT was added 30 min after the start of the reaction, allowing for the measurement of the stability of interactions already formed at the active site of urease (enzyme reactivation). In this case, enzymatic activity could be fully (100% for 14f) or partially (27.3–76.3% for other derivatives) recovered. The inhibition with phosphonic acids 1e–h appeared to be more susceptible to reconstitution than that achieved with the corresponding phosphonate esters (14e–h). Enzyme reactivation was also performed in an experiment where the enzymes were incubated with inhibitors for 2 h before the addition of substrate and measurement. Recovery in activity was significantly less pronounced and was observed only for compounds 14f, 1f, and 1h, which were relatively weaker inhibitors (Table).
2: DTT-Mediated S. pasteurii Urease Protection or Recovery from Inhibition
Molecular Modeling
Molecular modeling demonstrated a regular mode of binding of diethyl phosphonate inhibitors to bacterial ureases. For the enzyme from S. pasteurii, this involves the assumed opening of the isoselenazolone ring and the formation of the covalent S–Se adduct with the Cys322 residue, which generally corresponds to what was previously reported for Ebselen? and Ebselen-based inhibitors. ?,? Despite differences in the structure of the 2-substituent, representatives of alkyl (14a), aryl (14f), or extended alkylaryl compounds (14l) fit the active site similarly (Figure). The fit includes the incorporation of lipophilic portions of the inhibitors within the hydrophobic cleft formed by Met318, Met367, Ala366, and Ala170. The ethyl analogue 14a is buried somewhat deeper than the others (Figure, panel A), while the rigid p-phenyl derivative 14f is less favorably positioned despite having a large surface area for potential contacts (Figure, panel B). The 14l xylyl compound is the most extended, yet it fills the cavity tightly due to increased flexibility and a gently twisted conformation (Figure, panel C). In all cases, a phosphonate group points toward Arg339, allowing the formation of a hydrogen bond between the phosphoryl oxygen atom and the NH of the guanidino group. Conversely, this interaction prevents the guanidino group from engaging in hydrogen bonding with the oxygen atom of the ligand’s amide group, which is typical for Ebselen and its nonphosphorylated derivatives. ?−? ? This difference may explain the lack of improvement in antiurease activity observed with the phosphonate functionalization compared with the unmodified structure. Finally, one of the ethyl residues of the phosphonate ester is situated near lipophilic residues Leu319 and Phe335.
Modeled covalent complexes of inhibitors 14a (panel A), 14f (panel B), and 14l (panel C) with S. pasteurii urease (PDB id 5G4H). The enzyme is depicted as a solid ribbon with a solvent-accessible surface, while nickel ions are represented as blue spheres.
The predicted mode of interaction for inhibitors 14a, 14f, and 14l with H. pylori urease (see Figure S3) resembles that described for S. pasteurii. This is not surprising, as microbial ureases exhibit a high degree of regularity in the arrangement of the catalytic site region. ?,? These minor variations in the generally well-conserved mode of action of diethyl phosphonates with the enzymes rendered a more comprehensive discussion of the structure–activity relationship based on a molecular modeling complex. Besides conformational aspects of ligand–protein binding, other factors could influence inhibitor affinity, such as differences in the chemical reactivity of 2-alkyl versus 2-aryl and 2-benzylic benzisoselenazolones with the enzyme thiols.
The rational comments regarding the unexpectedly low potency of phosphonic acid 1a–h were also problematic. The simultaneous presence of the two functional fragments in these structures appeared nonoptimal and did not yield the expected synergy of action (Figure for S. pasteurii urease and Figure S4 for H. pylori urease). First, only alkylphosphonic acids 1a–d could bind to the nickel ions present in the active sites, with the optimal distance found for the shortest homologue 1a (Figures and S4, panel A). The longest aliphatic homologue 1d forms more constrained complexes; however, its structure offers an extended surface of lipophilic contacts (Figures and S4, panel B). Other benzisoselenazolone analogues, phenyl- (e.g., 1f; Figures and S4, panel C) or benzylphosphonic acids, could not locate the acidic functionality in proximity to the metal ions due to the spatial size and rigidity of their structures. Instead, the phosphorus-containing substituents of these compounds are buried deeper in the hydrophobic environment of the active site clefts, with the phosphonic acid group in the proximity of the imidazole of His323/His322. Second, these compounds do not form any specific hydrogen bonds that typically involve NH of the guanidino group of Arg339 (S. pasteurii urease) bonded either with the amide oxygen atom of Ebselen? or the phosphoryl oxygen atom of compounds 14a–l (vide supra). Concluding, the structures of the complexes of phosphonic acids with ureases indicated the lack of concerted mixed-type interactions indispensable for favorable binding.
Modeled covalent complexes of inhibitors 1a (panel A), 1d (panel B), and 1f (panel C) with S. pasteurii urease (PDB id 5G4H). The enzyme is depicted as a solid ribbon with a solvent-accessible surface, while nickel ions are represented as blue spheres.
Antimicrobial Activity
The antimicrobial activity of the selected potent inhibitors of whole cell ureolysis (five phosphonate esters 14e–h and 14k and phosphonic acid 1h) was further characterized on the H. pylori Tx30a strain. The antibacterial effect was moderate: the MIC values varied from 20 to 80 μM (8.24–33.0 μg/mL), were several fold higher than those previously reported for Ebselen, ?,? and approximately three orders of magnitude higher than that of amoxicillin, which was used as a positive control (Table). The most significant activity was observed for the best inhibitor 14f. Interestingly, ester 14h and corresponding acid 1h were found to be equipotent.
3: Antimicrobial Effects of Hybrid Organophosphorus/Selenium Urease Inhibitors, Compared to Amoxicillin, against H. pylori Tx30a
Furthermore, the potential of the phosphonate with the highest antibacterial activity (14f) to work in combination with the most common anti-H. pylori antibiotic (amoxicillin) was confirmed. Using checkerboard assays, an additive interaction between these compounds was shown by obtaining a fractional inhibitory concentration index (FICI) of 0.75 (Table). Interestingly, this value resulted from simultaneously halving the concentration of phosphonate 14f and reducing amoxicillin by a factor of 4 or vice versa. This suggests that each compound operates through a different antibacterial mechanism against H. pylori. On the one hand, amoxicillin might facilitate the entry of phosphonate 14f into H. pylori cells by weakening the cell envelope’s integrity. ?,? On the other hand, compound 14f, as an Ebselen derivative, could inhibit the antioxidant function of some H. pylori proteins, thereby enhancing the antibacterial effects of free oxygen radicals produced as a side effect of amoxicillin activity. ?,?,?
4: Antimicrobial Effects of Diethyl Phosphonate 14f in Combination with Amoxicillin against H. pylori Tx30a
Cytotoxicity
Finally, selected benzisoselenazolone inhibitors, six phosphonate esters (14a, 14d, and 14e–h) and one phosphonic acid (1h), were evaluated for their cytotoxic effects on eukaryotic cells, specifically normal fibroblasts from mouse embryo (BALB/3T3-L1) and epithelial cells from human embryo kidney (HEK-293). All compounds displayed moderate to low antiproliferative activity, with IC_50_ values typically ranging from 40 to 80 μM and in two cases exceeding 100 μM (Table). No substantial differences in growth inhibition were noted between benzisoselenazolones with different 2-substituent structures, specifically alkyl (14a and 14d) versus phenyl (14e and 14f) and benzyl (14g and 14h), or between an ester (14h) and an acid (1h), for both BALB/3T3-L1 and HEK-293 cells. The organoselenium/phosphorus inhibitors were significantly less toxic than cisplatin, which was used as a positive control, by an order of magnitude for murine fibroblasts and more than two orders of magnitude for human kidney cells.
5: Antiproliferative Activity of Selected Urease Inhibitors against Murine Embryonic Fibroblast Balb/3T3-L1 and Human Embryonic Kidney HEK-293 Cell Lines (IC50 Concentrations and Their 95% Confidence Intervals)
Summary
The phosphonic acid group is a prominent pharmacophoric functionality in numerous antiviral, antimicrobial, and anticancer drugs.? Regarding the mode of action, this group enhances the specific molecular recognition of bioactive compounds by target receptors, which frequently involves complexation with metal ions. In applying this general rationale to the present work, we combined the benzisoselenazolone structurecharacterized by its strong antiureolytic activitywith the phosphonate functionality, which is designed to bind to the nickel ions present in the enzyme’s active site. We expected an enhancement in the potency of the hybrid compounds; however, the effect of substitution was not cooperative. The inhibitory activity of phosphonic acids 1a–h against the model urease of S. pasteurii varied within a relatively narrow range (K i = 108–436 nM), indicating that the structural diversity of the phosphonoalkyl/aryl portion barely influenced enzyme–inhibitor interactions. The differentiation of the SAR data was much more pronounced for diethyl phosphonates 14a–l (K i = 5.06–453 nM) and involved almost two orders of magnitude between the most potent extended arylalkyl analogues and the phenyl derivatives of the lowest affinity (despite the absence of significant variations in the binding mode illustrated by molecular modeling). Apparently, none of the representatives from the two series of compounds outscored the activity of Ebselen, the prototypical benzisoselenazolone inhibitor of bacterial urease (K i* = 2.11 nM for S. pasteurii; Figure).?
Comparison of inhibitory activity of Ebselen and its para-diethyl phosphonate analogue 14f against urease of S. pasteurii and ureolysis of live cells of H. pylori.
Although phosphorylation seemed to deteriorate the inhibition of a purified bacterial enzyme from S. pasteurii, it significantly improved the prevention of ureolysis exhibited by pathogenic H. pylori cells. Phosphonic acids 1a–h were again less potent (IC_50_ = 187–2106 nM), which was expected due to the negative charge(s) that make the functional group very polar and poorly bioavailable because of inadequate permeability through microbial cell walls. To mitigate these unfavorable physicochemical properties, the polarity of phosphonic acids is often masked in prodrug forms using various protecting groups.? Ethyl esters are generally not applied for constructing prodrugs (not sufficiently cleavable), but in our case, they served a protective role. The structure of diethyl phosphonate enabled us to achieve unprecedented antiureolytic activity of compounds 14a–l (IC_50_ = 29.5–299 nM), which has never been reported for intact H. pylori cells. All developed compounds exhibited higher potency than Ebselen (IC_50_ = 495 nM; Figure), with IC_50_ values 12–17-fold lower for compounds 14e, 14f, and 14j–l. This effect was clearly the result of improved physicochemical properties combined with high complementarity to the target enzyme: the appropriate size and lipophilicity of the inhibitors balanced with the ability to form a specific hydrogen bond (differing from the binding postulated for Ebselen). The excellent antiureolytic activity in cells, advantageous bioavailability, and reasonable cytotoxicity make the developed compounds promising adjuvants in combination therapies against H. pylori infections, aimed at eliminating the major bacterial virulence factor. A cooperative antibacterial action of diethyl phosphonate 14f with amoxicillin was preliminarily demonstrated by decreasing the MIC values when both compounds were used simultaneously against H. pylori; however, understanding the molecular mechanisms governing these effects requires further investigation.
Experimental Section
Chemistry
General Methods
All reagents used were purchased from the following commercial suppliers: Merck Poland–Sigma-Aldrich, Avantor Performance Materials Poland, Chemland Poland, and Stanlab Poland. They were of analytical grade and used without further purification unless otherwise stated. Anhydrous dichloromethane was prepared prior to use by distillation over phosphorus pentoxide. Triethylamine was distilled and stored in potassium hydroxide. Microwave-induced reactions were performed on an Initiator^+^ SP Wave Biotage apparatus. Reactions were monitored by thin-layer chromatography (TLC) on 0.25 mm silica gel plates with fluorescent labels (silica gel 60F254, Merck), and components were visualized using UV light absorption and/or incubation with iodine. Flash chromatography was performed on a Teledyne ISCO CombiFlash using RediSep Gold silica or C18 columns. High-performance liquid chromatography was conducted on a Shimadzu system with the ReproSil-Pur Basic-C18 column from Maisch, SHIM-POL. The melting points were determined on an Electrothermal IA 91100 digital melting point apparatus using the standard open capillary method. The ^1^H, ^13^C, ^31^P and ^77^Se NMR spectra were recorded in CD_3_OD or D_2_O on a Jeol ECZ 400S spectrometer at frequencies of 399.8 MHz (^1^H), 151.0 or 100.5 MHz (^13^C), 161.8 MHz (^31^P), and 76.2 MHz (^77^Se), respectively, at 295 K. Chemical shifts were reported in parts per million (ppm, δ) downfield from tetramethylsilane. Residual solvent central signals were recorded as follows: CD_3_OD, δ_H_ = 3.31 ppm, δ_C_ = 49.00 ppm, and D_2_O, δ_H_ = 4.79 ppm. Coupling patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), quintet (quin), and multiplet (m). High-resolution mass spectra (HRMS) were recorded by using an electron spray ionization (ESI) technique on a Waters LCT Premier XE spectrometer. Analytical reversed-phase high-performance liquid chromatography was performed using the UFLC Shimadzu system and the CHROMSHELL C18-XB HPLC column, 4.6 × 75 mm (0 min, 10% B → 1 min, 10% B → 12.5 min, 90% B → 15 min, 90% B → 17 min, 10% B, flow rate 0.9 mL/min for compounds 14a–l, 1d–h, 15a, and 15b, or 0 min, 0% B → 10 min, 40% B → 13 min, 90% B → 14 min, 90% B → 15 min, 0% B, flow rate 0.9 mL/min for compounds 1a–c). Solvent A is 0.1% TFA in water; solvent B is 0.1% TFA in acetonitrile. Chromatograms were recorded at wavelengths of 222 and 254 nm using background compensation. The final 2-benzyl-1,2-benzisoselenazol-3(2H)-ones 14a–l, 1d–h, 15a, and 15b gave satisfactory NMR and HRMS spectra and were *>*95% pure.
Diethyl Phosphonates, Derivatives of 2-Substituted 1,2-Benzisoselenazol-3(2H)-ones (14a–l)
Compounds 14a–l were synthesized through the aminolysis of 2-(chloroseleno)benzoyl chloride (13), obtained from anthranilic acid (12), with diethyl aminophosphonates 5a–l (detailed procedures for their preparation are described in the Supporting Information), as outlined in the procedure previously reported with minor modifications. ?,? Anhydrous triethylamine (2.5 equiv) and 2-(chloroseleno)benzoyl chloride (13, 1 eq., dissolved in anhydrous dichloromethane, typically 2–5 mmol in 1 mL) were then added through a septum to a solution of a diethyl aminophosphonate 5a–l (1.0 equiv) in anhydrous dichloromethane (5 mL). After the mixture was stirred at room temperature for 48 h, a 5% aqueous sodium bicarbonate solution (20 mL) was added, and the product was extracted with methylene chloride (50 mL). The organic phase was washed with a 5% aqueous sodium bisulfate solution (20 mL) and brine (20 mL) and then dried over anhydrous Na_2_SO_4_. The drying agent was filtered off, and the filtrate was concentrated in vacuo. The residue was purified by flash chromatography (using a gradient of dichloromethane/methanol, 100/0 → 90/10 vv).
Diethyl 2-(3-oxobenzo[d][1,2]selenazol-2(3H)-yl)ethylphosphonate (14a)
Light orange solid, yield 63%, mp 123–124 °C. ^1^H NMR (400 MHz, CD_3_OD) δ 7.95–7.92 (m, 2H), 7.62 (ddd, J HH = 8.3, 7.2, 1.4 Hz, 1H), 7.45 (ddd, J HH = 8.0, 7.2, 1.0 Hz, 1H), 4.15–4.01 (m, 6H), 2.35–2.26 (m, 2H), 1.29 (t, J HH = 7.1 Hz, 6H). ^13^C NMR (101 MHz, CD_3_OD) δ 169.35, 141.21, 133.18, 128.75, 128.73, 127.20, 126.40, 63.54 (d, J CP = 6.6 Hz), 39.84 (d, J CP = 2.1 Hz), 26.66 (d, J CP = 138.9 Hz), 16.62 (d, J CP = 6.2 Hz). ^31^P NMR (162 MHz, CD_3_OD) δ 28.88. ^77^Se NMR (76 MHz, CD_3_OD) δ 914.51. HRMS (ESI) m/z calculated for C_13_H_18_NO_4_PSe+H^+^ 364.0217, found 364.0226.
Diethyl 3-(3-oxobenzo[d][1,2]selenazol-2(3H)-yl)propylphosphonate (14b)
Brown oil, yield 68%. ^1^H NMR (400 MHz, CD_3_OD) δ 7.95–7.92 (m, 2H), 7.61 (ddd, J HH = 8.3, 7.2, 1.4 Hz, 1H), 7.44 (ddd, J HH = 8.0, 7.2, 1.0 Hz, 1H), 4.13–4.03 (m, 4H), 3.90 (t, J HH = 6.8 Hz, 2H), 2.03–1.94 (m, 2H), 1.91–1.82 (m, 2H), 1.30 (t, J HH = 7.1 Hz, 6H). ^13^C NMR (101 MHz, CD_3_OD) δ 169.39, 140.93, 133.10, 128.85, 128.82, 127.21, 126.36, 63.30 (d, J CP = 6.6 Hz), 45.40 (d, J CP = 19.0 Hz), 24.45 (d, J CP = 4.7 Hz), 23.07 (d, J CP = 142.2 Hz), 16.71 (d, J CP = 6.0 Hz). ^31^P NMR (162 MHz, CD_3_OD) δ 32.83. ^77^Se NMR (76 MHz, CD_3_OD) δ 907.96. HRMS (ESI) m/z calculated for C_14_H_20_NO_4_PSe+H^+^ 378.0373, found 378.0396.
Diethyl 4-(3-oxobenzo[d][1,2]selenazol-2(3H)-yl)butylphosphonate (14c)
Brown oil, yield 73%. ^1^H NMR (400 MHz, CD_3_OD) δ 7.95–7.92 (m, 2H), 7.62 (ddd, J HH = 8.2, 7.2, 1.4 Hz, 1H), 7.45 (ddd, J HH = 8.7, 7.2, 1.0 Hz, 1H), 4.13–4.01 (m, 4H), 3.88 (t, J HH = 6.8 Hz, 2H), 1.92–1.82 (m, 4H), 1.68–1.61 (m, 2H), 1.27 (t, J HH = 7.1 Hz, 6H). ^13^C NMR (101 MHz, CD_3_OD) δ 169.31, 140.89, 133.03, 129.00, 128.82, 127.19, 126.33, 63.15 (d, J CP = 6.6 Hz), 44.46, 31.86 (d, J CP = 16.2 Hz), 25.19 (d, J CP = 140.5 Hz), 20.42 (d, J CP = 5.1 Hz), 16.69 (d, J CP = 6.1 Hz). ^31^P NMR (162 MHz, CD_3_OD) δ 33.56. ^77^Se NMR (76 MHz, CD_3_OD) δ 902.97. HRMS (ESI) m/z calculated for C_15_H_22_NO_4_PSe+H^+^ 392.0530, found 392.0533.
Diethyl 5-(3-oxobenzo[d][1,2]selenazol-2(3H)-yl)pentylphosphonate (14d)
Brown oil, yield 81%. ^1^H NMR (400 MHz, CD_3_OD) δ 7.94–7.91 (m, 2H), 7.61 (ddd, J HH = 8.2, 7.2, 1.4 Hz, 1H), 7.44 (ddd, J HH = 7.9, 7.2, 1.1 Hz, 1H), 4.09–4.01 (m, 4H), 3.84 (t, J HH = 7.0 Hz, 2H), 1.83–1.72 (m, 4H), 1.67–1.59 (m, 2H), 1.52–1.44 (m, 2H), 1.29 (t, J HH = 7.0 Hz, 6H). ^13^C NMR (101 MHz, CD_3_OD) δ 169.23, 140.87, 132.99, 129.08, 128.80, 127.17, 126.30, 63.09 (d, J CP = 6.6 Hz), 45.14, 30.91, 28.39 (d, J CP = 16.1 Hz), 25.62 (d, J CP = 140.4 Hz), 23.09 (d, J CP = 5.2 Hz), 16.71 (d, J CP = 6.0 Hz). ^31^P NMR (162 MHz, CD_3_OD) δ 33.90. ^77^Se NMR (76 MHz, CD_3_OD) δ 902.95. HRMS (ESI) m/z calculated for C_16_H_24_NO_4_PSe+H^+^ 406.0687, found 406.0688.
Diethyl 3-(3-oxobenzo[d][1,2]selenazol-2(3H)-yl)phenylphosphonate (14e)
Light yellow crystals, yield 53%, mp 107–110 °C. ^1^H NMR (400 MHz, CD_3_OD) δ 8.09–7.98 (m, 3H), 7.88 (ddd, J HH = 7.8, 1.2 Hz, J HP = 2.3 Hz, 1H), 7.73–7.64 (m, 3H), 7.50 (ddd, J = 8.1, 7.3, 1.1 Hz, 1H), 4.20–4.12 (m, 4H), 1.35 (t, J HH = 7.0 Hz, 6H). ^13^C NMR (101 MHz, CD_3_OD) δ 166.82, 139.93 (d, J CP = 18.9 Hz), 139.37, 132.58, 129.80 (d, J CP = 16.5 Hz), 129.41 (d, J CP = 3.4 Hz), 129.22 (d, J CP = 9.4 Hz), 129.02 (d, J CP = 189.9 Hz), 128.19, 127.95, 127.83, 126.35, 125.03, 62.79 (d, J CP = 5.9 Hz), 15.31 (d, J CP = 6.3 Hz). ^31^P NMR (162 MHz, CD_3_OD) δ 18.30. ^77^Se NMR (76 MHz, CD_3_OD) δ 969.96. HRMS (ESI) m/z calculated for C_17_H_18_NO_4_PSe+H^+^ 412.0217, found 412.0180.
Diethyl 4-(3-oxobenzo[d][1,2]selenazol-2(3H)-yl)phenylphosphonate (14f)
Light yellow crystals, yield 74%, mp 165–170 °C. ^1^H NMR (400 MHz, CD_3_OD) δ 8.02–7.97 (m, 2H), 7.88–7.85 (m, 4H), 7.69 (ddd, J HH = 8.3, 7.2, 1.4 Hz, 1H), 7.50 (ddd, J HH = 8.1, 7.2, 1.0 Hz, 1H), 4.19–4.08 (m, 4H), 1.34 (t, J HH = 7.1 Hz, 6H). ^13^C NMR (101 MHz, CD_3_OD) δ 168.11, 145.23, 140.57, 134.02, 133.91, 129.56, 129.40, 127.68, 126.29, 126.14 (d, J CP = 193.3 Hz), 125.84 (d, J CP = 15.5 Hz), 63.92 (d, J CP = 5.7 Hz), 16.62 (d, J CP = 6.3 Hz). ^31^P NMR (162 MHz, CD_3_OD) δ 19.05. ^77^Se NMR (76 MHz, CD_3_OD) δ 968.38. HRMS (ESI) m/z calculated for C_17_H_18_NO_4_PSe+H^+^ 412.0217, found 412.0218.
Diethyl 3-(3-oxobenzo[d][1,2]selenazol-2(3H)-yl)benzylphosphonate (14g)
Yellow oil, yield 70%. ^1^H NMR (400 MHz, CD_3_OD) δ 8.01–7.97 (m, 2H), 7.68 (ddd, J HH = 8.3, 7.2, 1.4 Hz, 1H), 7.57–7.48 (m, 3H), 7.44–7.40 (m, 1H), 7.28 (ddd, J HH = 7.8, 1.3 Hz, J HP = 2.8 Hz, 1H), 4.13–4.04 (m, 4H), 3.32 (d, J HP = 21.8 Hz, 2H), 1.29 (t, J HH = 7.1 Hz, 6H). ^13^C NMR (101 MHz, CD_3_OD) δ 167.97, 140.95, 140.64 (d, J CP = 3.3 Hz), 134.42 (d, J CP = 9.3 Hz), 133.65, 130.45 (d, J CP = 3.1 Hz), 129.58 (d, J CP = 6.6 Hz), 129.39, 129.29, 128.12 (d, J CP = 6.5 Hz), 127.53, 126.27, 125.34 (d, J CP = 3.6 Hz), 63.85 (d, J CP = 6.9 Hz), 33.48 (d, J CP = 137.8 Hz), 16.72 (d, J CP = 6.0 Hz). ^31^P NMR (162 MHz, CD_3_OD) δ 27.61. ^77^Se NMR (76 MHz, CD_3_OD) δ 969.06. HRMS (ESI) m/z calculated for C_18_H_20_NO_4_PSe+H^+^ 426.0374, found 426.0385.
Diethyl 4-(3-oxobenzo[d][1,2]selenazol-2(3H)-yl)benzylphosphonate (14h)
Light yellow crystals, yield 66%, mp 140–142 °C. ^1^H NMR (400 MHz, CD_3_OD) δ 8.01–7.97 (m, 2H), 7.68 (ddd, J HH = 8.3, 7.2, 1.4 Hz, 1H), 7.57 (dd, J HH = 8.6 Hz, J HP = 1.1 Hz, 2H), 7.50 (ddd, J HH = 7.5, 7.3, 1.0 Hz, 1H), 7.42 (dd, J HH = 8.5 Hz, J HP = 2.6 Hz, 2H), 4.12–4.04 (m, 4H), 3.30 (d, J HP = 21.7 Hz, 2H), 1.29 (t, J HH = 7.1 Hz, 6H). ^13^C NMR (101 MHz, CD_3_OD) δ 167.96, 140.91, 139.35, 133.63, 131.89 (d, J CP = 6.6 Hz), 131.70 (d, J CP = 9.5 Hz), 129.38, 129.30, 127.52, 126.67 (d, J CP = 3.2 Hz), 126.25, 63.78 (d, J CP = 6.9 Hz), 33.22 (d, J CP = 137.9 Hz), 16.68 (d, J CP = 6.0 Hz). ^31^P NMR (162 MHz, CD_3_OD) δ 27.71. ^77^Se NMR (76 MHz, CD_3_OD) δ 968.23. HRMS (ESI) m/z calculated for C_18_H_20_NO_4_PSe+H^+^ 426.0374, found 426.0365.
Diethyl 2-(3-(3-oxobenzo[d][1,2]selenazol-2(3H)-yl)phenyl)ethylphosphonate (14i)
Yellow oil, yield 56%. ^1^H NMR (400 MHz, CD_3_OD) δ 8.01–7.96 (m, 2H), 7.68 (ddd, J HH = 8.2, 7.3, 1.4 Hz, 1H), 7.52–7.48 (m, 2H), 7.42–7.38 (m, 2H), 7.25–7.22 (m, 1H), 4.12–4.05 (m, 4H), 2.99–2.91 (m, 2H), 2.22–2.13 (m, 2H), 1.30 (t, J HH = 7.1 Hz, 6H). ^13^C NMR (101 MHz, CD_3_OD) δ 167.98, 143.64 (d, J CP = 15.8 Hz), 141.01, 140.56, 133.61, 130.59, 129.39, 129.28, 128.02, 127.51, 126.67, 126.24, 124.84, 63.28 (d, J CP = 6.7 Hz), 29.34 (d, J CP = 4.6 Hz), 27.55 (d, J CP = 139.5 Hz), 16.71 (d, J CP = 6.1 Hz). ^31^P NMR (162 MHz, CD_3_OD) δ 32.28. ^77^Se NMR (76 MHz, CD_3_OD) δ 969.66. HRMS (ESI) m/z calculated for C_19_H_22_NO_4_PSe+H^+^ 440.0530, found 440.0538.
Diethyl 2-(4-(3-oxobenzo[d][1,2]selenazol-2(3H)-yl)phenyl)ethylphosphonate (14j)
Light yellow crystals, yield 61%, mp 137–139 °C. ^1^H NMR (400 MHz, CD_3_OD) δ 8.00–7.96 (m, 2H), 7.67 (ddd, J HH = 8.2, 7.2, 1.4 Hz, 1H), 7.52–7.47 (m, 3H), 7.37–7.34 (m, 2H), 4.12–4.05 (m, 4H), 2.96–1.89 (m, 2H), 2.17–2.11 (m, 2H), 1.29 (t, J HH = 7.1 Hz, 6H). ^13^C NMR (101 MHz, CD_3_OD) δ 167.99, 141.15 (d, J CP = 15.8 Hz), 140.99, 138.70, 133.58, 130.20, 129.36, 129.27, 127.51, 126.95, 126.25, 63.27 (d, J CP = 6.6 Hz), 29.06 (d, J CP = 4.6 Hz), 27.65 (d, J CP = 139.2 Hz), 16.72 (d, J CP = 6.1 Hz). ^31^P NMR (162 MHz, CD_3_OD) δ 32.30. ^77^Se NMR (76 MHz, CD_3_OD) δ 968.14. HRMS (ESI) m/z calculated for C_19_H_22_NO_4_PSe+H^+^ 440.0530, found 440.0523.
Diethyl 3-((3-oxobenzo[d][1,2]selenazol-2(3H)-yl)methyl)benzylphosphonate (14k)
Yellow oil, yield 52%. ^1^H NMR (400 MHz, CD_3_OD) δ 7.97(ddd, J HH = 7.9, 1.4, 0.6 Hz, 1H), 7.89 (ddd, J HH = 8.1, 0.9, 0.9 Hz, 1H), 7.61 (ddd, J HH = 8.3, 7.2, 1.4 Hz, 1H), 7.46 (ddd, J HH = 8.1, 7.2, 1.0 Hz, 1H), 7.39–7.23 (m, 4H), 4.99 (s, 2H), 4.03–3.93 (m, 4H), 3.23 (d, J HP = 21.8 Hz, 2H), 1.17 (t, J HH = 7.1 Hz, 6H). ^13^C NMR (101 MHz, CD_3_OD) 169.21, 141.14, 139.37 (d, J CP = 3.3 Hz), 133.54 (d, J CP = 9.4 Hz), 133.14, 130.78 (d, J CP = 6.5 Hz), 129.99 (d, J CP = 3.2 Hz), 128.98, 128.89, 128.01, 127.97, 127.21, 126.33, 63.75 (d, J CP = 6.9 Hz), 54.81, 33.61 (d, J CP = 137.7 Hz), 16.61 (d, J CP = 6.0 Hz). ^31^P NMR (162 MHz, CD_3_OD) δ 27.84. ^77^Se NMR (76 MHz, CD_3_OD) 903.92. HRMS (ESI) m/z calculated for C_19_H_22_NO_4_PSe+H^+^ 440.0530, found 440.0538.
Diethyl 4-((3-oxobenzo[d][1,2]selenazol-2(3H)-yl)methyl)benzylphosphonate (14l)
Light yellow crystals, yield 56%, mp 152–153 °C. ^1^H NMR (400 MHz, CD_3_OD) δ 7.96 (ddd, J HH = 7.8, 1.4, 0.6 Hz, 1H), 7.89 (ddd, J HH = 8.1, 0.9, 0.9 Hz, 1H), 7.61 (ddd, J HH = 8.2, 7.2, 1.4 Hz, 1H), 7.45 (ddd, J HH = 8.1, 7.2, 1.0 Hz, 1H), 7.33–7.31 (m, 4H), 4.98 (d, J HP = 1.7 Hz, 2H), 4.06–3.97 (m, 4H), 3.24 (d, J HP = 21.7 Hz, 2H), 1.24 (t, J HH = 7.1 Hz, 6H). ^13^C NMR (101 MHz, CD_3_OD) δ 169.23, 141.11, 137.76 (d, J CP = 3.9 Hz), 133.12, 132.72 (d, J CP = 9.3 Hz), 131.43 (d, J CP = 6.6 Hz), 129.53 (d, J CP = 3.2 Hz), 128.94, 128.91, 127.21, 126.31, 63.74 (d, J CP = 6.9 Hz), 54.81, 33.40 (d, J CP = 137.9 Hz), 16.65 (d, J CP = 6.0 Hz). ^31^P NMR (162 MHz, CD_3_OD) δ 27.97. ^77^Se NMR (76 MHz, CD_3_OD) 903.24. HRMS (ESI) m/z calculated for C_19_H_22_NO_4_PSe+H^+^ 440.0530, found 440.0541.
Phosphonic Acids, Derivatives of 2-Substituted 1,2-Benzisoselenazol-3(2H)-ones (1a–h)
Compounds 1a–h were obtained through the microwave-heated acidic hydrolysis of 14a–h. An ester 14a–h (1.0 mmol) dissolved in acetic acid (1 mL) and 36–38% hydrochloric acid (1 mL) was heated in a microwave oven at 140 °C for 20 min. After the mixture was cooled to room temperature and volatiles evaporated in vacuo, the residue was purified by reverse-phase HPLC chromatography (using a gradient of acetonitrile/water, 10/90 → 90/10 vv + 0.05% TFA).
2-(3-Oxobenzo[d][1,2]selenazol-2(3H)-yl)ethylphosphonic acid (1a)
White solid, yield 59%, mp 215–217 °C. ^1^H NMR (400 MHz, D_2_O) δ 7.80–7.77 (m, 2H), 7.60 (ddd, J HH = 8.4, 7.2, 1.3 Hz, 1H), 7.44 (ddd, J HH = 8.0, 7.2, 1.1 Hz, 1H), 3.98–3.93 (m, 2H), 1.99–1.89 (m, 2H). ^13^C NMR (101 MHz, D_2_O) δ 167.98, 137.43, 133.05, 127.88, 127.22, 126.12, 125.09, 44.29 (d, J CP = 7.2 Hz), 27.02 (d, J CP = 130.7 Hz). ^31^P NMR (162 MHz, D_2_O) δ 17.16. ^77^Se NMR (76 MHz, D_2_O) δ 931.78. HRMS (ESI) m/z calculated for C_9_H_10_NO_4_PSe+H^+^ 307.9591, found 307.9597.
3-(3-Oxobenzo[d][1,2]selenazol-2(3H)-yl)propylphosphonic acid (1b)
White solid, yield 63%, mp 222–223 °C. ^1^H NMR (400 MHz, D_2_O) δ 7.75–7.73 (m, 2H), 7.57 (ddd, J HH = 8.5, 7.2, 1.3 Hz, 1H), 7.39 (ddd, J HH = 8.0, 7.2, 1.0 Hz, 1H), 3.78 (t, J HH = 7.1 Hz, 2H), 1.94–1.84 (m, 2H), 1.50–1.42 (m, 2H). ^13^C NMR (101 MHz, D_2_O) δ 168.37, 139.43, 132.19, 127.31, 126.76, 126.22, 124.50, 46.08 (d, J CP = 19.8 Hz), 25.96 (d, J CP = 131.7 Hz), 25.13 (d, J CP = 3.6 Hz). ^31^P NMR (162 MHz, D_2_O) δ 22.77. ^77^Se NMR (76 MHz, D_2_O) δ 925.64. HRMS (ESI) m/z calculated for C_10_H_12_NO_4_PSe+H^+^ 321.9748, found 321.9755.
4-(3-Oxobenzo[d][1,2]selenazol-2(3H)-yl)butylphosphonic acid (1c)
White solid, yield 66%, mp 226–228 °C. ^1^H NMR (400 MHz, D_2_O) δ 7.78–7.73 (m, 2H), 7.55 (ddd, J HH = 8.2, 7.2, 1.4 Hz, 1H), 7.41–7.37 (m, 1H), 3.70 (t, J HH = 7.4 Hz, 2H), 1.72 (quin, J HH = 7.4 Hz, 2H), 1.56–1.50 (m, 2H), 1.44–1.35 (m, 2H). ^13^C NMR (101 MHz, D_2_O) δ 168.51, 138.19, 132.08, 127.89, 127.30, 126.32, 125.49, 44.23, 31.49 (d, J CP = 17.2 Hz), 29.02 (d, J CP = 130.5 Hz), 21.56 (d, J CP = 3.8 Hz). ^31^P NMR (162 MHz, D_2_O) δ 23.38. ^77^Se NMR (76 MHz, D_2_O) δ 923.75. HRMS (ESI) m/z calculated for C_11_H_14_NO_4_PSe+H^+^ 335.9904, found 335.9909.
5-(3-Oxobenzo[d][1,2]selenazol-2(3H)-yl)pentylphosphonic acid (1d)
White solid, yield 70%, mp 190–192 °C. ^1^H NMR (400 MHz, D_2_O) δ 7.72–7.69 (m, 2H), 7.54 (ddd, J HH = 8.3, 7.2, 1.3 Hz, 1H), 7.36 (ddd, J HH = 8.0, 7.2, 1.0 Hz, 1H), 3.70 (t, J HH = 7.1 Hz, 2H), 1.67 (quin, J HH = 7.3 Hz, 2H), 1.55–1.29 (m, 6H). ^13^C NMR (101 MHz, D_2_O) δ 168.22, 139.34, 132.11, 127.26, 126.79, 126.18, 124.45, 44.92, 29.42, 28.88 (d, J CP = 131.4 Hz), 27.73 (d, J CP = 17.8 Hz), 23.50 (d, J CP = 4.1 Hz). ^31^P NMR (162 MHz, D_2_O) δ 24.35. ^77^Se NMR (76 MHz, D_2_O) δ 923.22. HRMS (ESI) m/z calculated for C_12_H_16_NO_4_PSe+H^+^ 350.0061, found 350.0070.
3-(3-Oxobenzo[d][1,2]selenazol-2(3H)-yl)phenylphosphonic acid (1e)
White solid, yield 60%, mp 223–225 °C. ^1^H NMR (400 MHz, D_2_O) δ 7.89–7.84 (m, 2H), 7.78–7.66 (m, 3H), 7.54–7.46 (m, 3H). ^13^C NMR (101 MHz, D_2_O) δ 167.95, 160.49, 142.71 (d, J CP = 165.6 Hz), 140.08, 136.96 (d, J CP = 15.7 Hz), 132.88, 130.11 (d, J CP = 8.2 Hz), 129.02 (d, J CP = 13.5 Hz), 128.10 (d, J CP = 9.5 Hz), 128.01, 126.82, 126.66 (t, J CP = 13.4 Hz), 124.63. ^31^P NMR (162 MHz, D_2_O) δ 10.37. ^77^Se NMR (76 MHz, D_2_O) δ 997.23. HRMS (ESI) m/z calculated for C_13_H_10_NO_4_PSe+H^+^ 355.9591, found 355.9593.
4-(3-Oxobenzo[d][1,2]selenazol-2(3H)-yl)phenylphosphonic acid (1f)
White solid, yield 65%, mp >260 °C. ^1^H NMR (400 MHz, D_2_O) δ 7.83–7.75 (m, 4H), 7.62 (ddd, J HH = 8.5, 7.2, 1.3 Hz, 1H), 7.47–7.41 (m, 3H). ^13^C NMR (101 MHz, D_2_O) δ 167.72, 162.00, 140.83 (d, J CP = 166.8 Hz), 139.89, 137.78 (d, J CP = 3.4 Hz), 132.81, 131.44 (d, J CP = 9.5 Hz), 127.90, 126.57, 126.37, 125.61 (d, J CP = 13,1 Hz), 124.49. ^31^P NMR (162 MHz, D_2_O) δ 10.84. ^77^Se NMR (76 MHz, D_2_O) δ 997.91. HRMS (ESI) m/z calculated for C_13_H_10_NO_4_PSe+H^+^ 355.9591, found 355.9673.
3-(3-Oxobenzo[d][1,2]selenazol-2(3H)-yl)benzylphosphonic acid (1g)
White solid, yield 48%, mp 200–205 °C. ^1^H NMR (400 MHz, D_2_O) δ 7.94–7.87 (m, 2H), 7.73–7.68 (m, 1H), 7.55–7.51 (m, 1H), 7.44–7.36 (m, 3H), 7.30 (ddd, J HH = 7.5, 1.9 Hz, J HP = 1.9 Hz, 1H), 2.90 (d, J HP = 19.6 Hz, 2H). ^13^C NMR (101 MHz, D_2_O) δ 168.04, 162.49, 140.33 (d, J CP = 7.9 Hz), 137.17, 132.89, 129.66 (d, J CP = 5.2 Hz), 129.14, 128.04, 127.58 (d, J CP = 5.6 Hz), 126.67, 126.60, 124.67, 123.41, 37.24 (d, J CP = 121.7 Hz). ^31^P NMR (162 MHz, D_2_O) δ 17.71. ^77^Se NMR (76 MHz, D_2_O) δ 996.60. HRMS (ESI) m/z calculated for C_14_H_12_NO_4_PSe+H^+^ 369.9747, found 369.9730.
4-(3-Oxobenzo[d][1,2]selenazol-2(3H)-yl)benzylphosphonic acid (1h)
White solid, yield 53%, mp >260 °C. ^1^H NMR (400 MHz, D_2_O) δ 7.81–7.76 (m, 2H), 7.64–7.60 (m, 1H), 7.45–7.33 (m, 5H), 2.89 (d, J HP = 19.8 Hz, 2H). ^13^C NMR (101 MHz, D_2_O) δ 167.73, 162.07, 139.93, 138.84, 134.57, 132.71, 130.54 (d, J CP = 5.5 Hz), 127.87, 126.56, 125.99, 124.50, 37.16 (d, J CP = 121.9 Hz). ^31^P NMR (162 MHz, D_2_O) δ 17.82. ^77^Se NMR (76 MHz, D_2_O) δ 996.14. HRMS (ESI) m/z calculated for C_14_H_12_NO_4_PSe+H^+^ 369.9747, found 369.9730.
Monoethyl Esters of Phosphonic Acids (15a and 15b)
To diethyl ester 14a or 14d (1.0 mmol) dissolved in methanol (2 mL) was added 2 M aqueous solution of NaOH (2 mL) was added. The mixture was stirred for 2 h, and volatiles were evaporated in vacuo. The residue was purified by reverse-phase HPLC chromatography (using a gradient of acetonitrile/water, 10/90 → 90/10 vv + 0.05% TFA).
Monoethyl Ester of 2-(3-oxobenzo[d][1,2]selenazol-2(3H)-yl)ethylphosphonic acid (15a)
White solid, yield 43%, mp 210–211 °C. ^1^H NMR (400 MHz, D_2_O) δ 7.80 (dd, J HH = 7.9, 1.2 Hz, 1H), 7.60 (dd, J HH = 7.7, 1.5 Hz, 1H), 7.36 (ddd, J HH = 8.8, 7.9, 1.5 Hz, 1H), 7.31 (ddd, J HH = 8.8, 7.7, 1.2 Hz, 1H), 3.86 (quin, J HH ∼ J HP ∼ 7.0 Hz, 2H), 3.66–3.59 (m, 2H), 2.00–1.92 (m, 2H), 1.16 (t, J HH = 7.1 Hz, 3H). ^13^C NMR (101 MHz, D_2_O) δ 169.54, 135.60, 132.08, 130.84, 128.66, 127.56, 126.58, 60.78 (d, J CP = 6.0 Hz), 37.95 (d, J CP = 4.0 Hz), 26.70 (d, J CP = 131.6 Hz), 15.89 (d, J CP = 6.6 Hz). ^31^P NMR (162 MHz, D_2_O) δ 24.10. ^77^Se NMR (76 MHz, D_2_O) δ 925.56. HRMS (ESI) m/z calculated for C_11_H_14_NO_4_PSe+H^+^ 335.9904, found 335.9968.
Monoethyl Ester of 5-(3-oxobenzo[d][1,2]selenazol-2(3H)-yl)pentylphosphonic acid (15d)
(P2.12.5) White solid, yield 48%, mp 182–183 °C. ^1^H NMR (400 MHz, D_2_O) δ 7.75 (dd, J HH = 7.9, 1.2 Hz, 1H), 7.61 (dd, J HH = 7.6, 1.6 Hz, 1H), 7.33 (ddd, J HH = 8.9, 7.9, 1.6 Hz, 1H), 7.28 (ddd, J HH = 8.9, 7.6, 1.2 Hz, 1H), 3.83–3.72 (m, 2H), 3.47 (t, J HH = 6.9 Hz, 2H), 1.60 (quin, J HH ∼ 7.0 Hz, 2H), 1.55–1.42 (m, 4H), 1.42–1.33 (m, 2H), 1.14 (t, J HH = 7.1 Hz, 3H). ^13^C NMR (101 MHz, D_2_O) δ 169.42, 135.89, 131.99, 130.81, 128.40, 127.53, 126.61, 60.37 (d, J CP = 5.7 Hz), 42.49, 28.76, 27.41 (d, J CP = 17.2 Hz), 25,99 (d, J CP = 134.1 Hz), 22.58 (d, J CP = 5.2 Hz), 15.97 (d, J CP = 6.3 Hz). ^31^P NMR (162 MHz, D_2_O) δ 29.26. ^77^Se NMR (76 MHz, D_2_O) δ 917.83. HRMS (ESI) m/z calculated for C_14_H_20_NO_4_PSe+H^+^ 378.0373, found 378.0499.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ureases. Functions, Classes, and Applications. In Foundations and Frontiers in Enzymology, 1st ed.; Ligabue-Braun, R. ; Carlini, C. R. , Eds.; Academic Press, 2024.
- 2Sumner J. B.The Isolation and Crystallization of the Enzyme Urease J. Biol. Chem.192669243544110.1016/S 0021-9258(18)84560-4 · doi ↗
- 3Dixon N. E.Gazzola C.Watters J. J.Blakeley R. L.Zerner B.Jack Bean Urease (EC 3.5.1.5). A Metalloenzyme. A Simple Biological Role for Nickel J. Am. Chem. Soc.197597144131413310.1021/ja 00847 a 0451159216 · doi ↗ · pubmed ↗
- 4Fowler D.Coyle M.Skiba U.Sutton M. A.Cape J. N.Reis S.Sheppard L. J.Jenkins A.Grizzetti B.Galloway J. N.Vitousek P.Leach A.Bouwman A. F.Butterbach-Bahl K.Dentener F.Stevenson D.Amann M.Voss M.The Global Nitrogen Cycle in the Twenty-First Century Philos. Trans. R. Soc., B 201336816212013016410.1098/rstb.2013.0164 PMC 368274823713126 · doi ↗ · pubmed ↗
- 5Motasim A. M.Samsuri A. W.Nabayi A.Akter A.Haque M. A.Abdul Sukor A. S.Adibah A. M.Urea Application in Soil: Processes, Losses, and AlternativesA Review Discovery Agric.202424210.1007/s 44279-024-00060-z · doi ↗
- 6Glibert P. M.Harrison J.Heil C.Seitzinger S.Escalating Worldwide Use of UreaA Global Change Contributing to Coastal Eutrophication Biogeochemistry 200677344146310.1007/s 10533-005-3070-5 · doi ↗
- 7Follmer C.Ureases as a Target for the Treatment of Gastric and Urinary Infections J. Clin. Pathol.201063542443010.1136/jcp.2009.07259520418234 · doi ↗ · pubmed ↗
- 8Flannigan R.Choy W. H.Chew B.Lange D.Renal Struvite StonesPathogenesis, Microbiology, and Management Strategies Nat. Rev. Urol.201411633334110.1038/nrurol.2014.9924818849 · doi ↗ · pubmed ↗