Refractory Digital Ulcers in Systemic Sclerosis Sine Scleroderma Associated With Antiphospholipid Syndrome: A Case-Based Review
Inês Almeida, Liliana Saraiva, Vera Romão, João Tavares, Inês Fróis Cunha, Maura Couto

TL;DR
A rare case of systemic sclerosis sine scleroderma with antiphospholipid syndrome is reported, where anticoagulation successfully treated severe digital ulcers.
Contribution
This is the first reported case of ssSSc with secondary APS presenting with recurrent digital ulcers and pulmonary embolism.
Findings
Anticoagulation led to complete healing of digital ulcers within two months.
The patient tested positive for lupus anticoagulant and anti-β2-glycoprotein I IgG, confirming secondary APS.
This case highlights the importance of considering APS in SSc patients with refractory digital ulcers.
Abstract
Digital ulcers (DUs) are a severe manifestation of the vasculopathy underlying systemic sclerosis (SSc). In cases refractory to treatment, the presence of other modifiable causes of vasculopathy, such as antiphospholipid syndrome (APS), should be considered, although its diagnosis in patients with systemic sclerosis sine scleroderma (ssSSc) is exceedingly rare. We report the case of a 20-year-old Caucasian female with ssSSc, who presented with Raynaud’s phenomenon (RP), recurrent DU, telangiectasias, inflammatory arthralgias, dyspepsia, and epigastric pain, in the absence of skin thickening. Laboratory investigation revealed a high-titre antinuclear antibody (ANA; 1:2560) with an anti-centromere pattern, and positivity for both anti-centromere antibodies (ACA) and anti-Ro-52 antibodies. Nailfold capillaroscopy showed an early scleroderma pattern. Despite optimized vasodilatory therapy,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
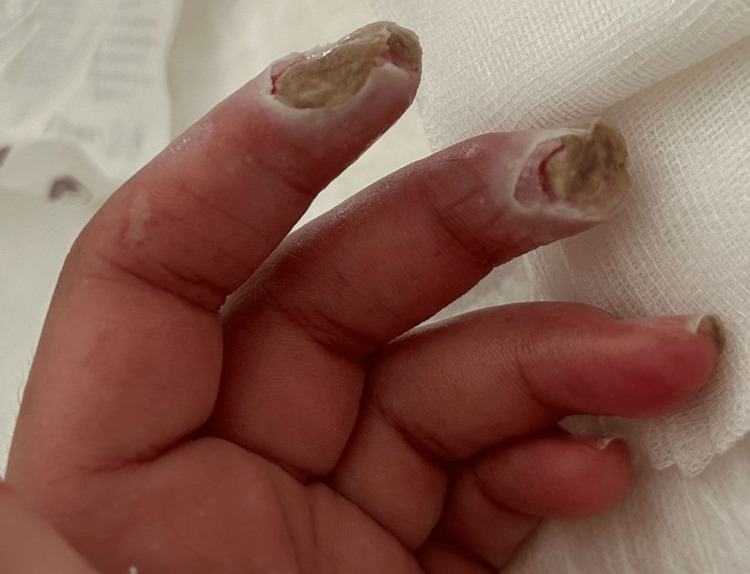 Figure 1
Figure 1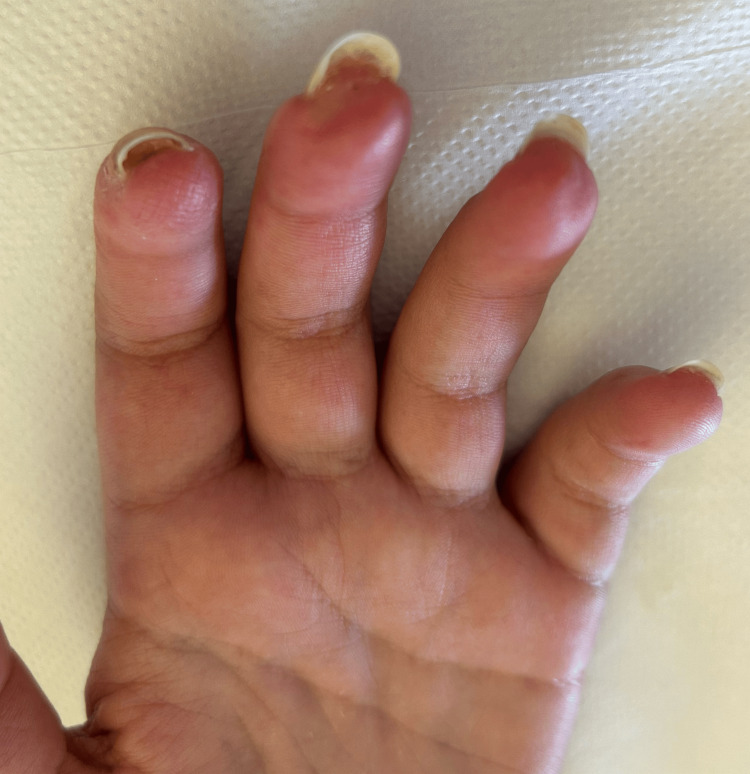 Figure 2
Figure 2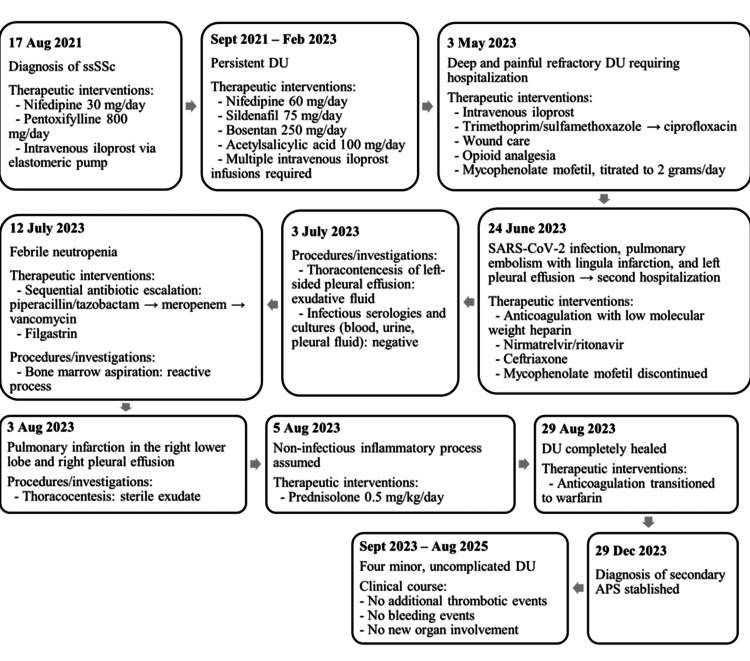 Figure 3
Figure 3| Investigation | Result | Reference range |
| ANA | 1:2560, centromere pattern | <1:160 |
| ACA | Positive | Negative |
| Anti-Ro-52 | Positive | Negative |
| LA | Positive | Negative |
| Anti-β2GPI IgG | 9.0 U/mL | 0.0-10.0 U/mL |
| Anti-β2GPI IgM | 4.0 U/mL | 0.0-10.0 U/mL |
| aCL IgG | 22.0 U/mL | 0.0-40.0 U/mL |
| aCL IgM | 5.3 U/mL | 0.0-40.0 U/mL |
| Investigation | Result | Reference range |
| Haemoglobin | 14.4 g/dL | 12.0-15.0 g/dL |
| White blood cells | 6.13 × 109/L | 4.50-11.50 × 109/L |
| Neutrophils | 3.3 × 109/L | 2.0-7.5 × 109/L |
| Platelet count | 177 × 109/L | 150.0-450.0 × 109/L |
| ESR | 24.0 mm/h | 0.0-20.0 mm/h |
| CRP | 0.71 mg/dL | <0.50 mg/dL |
| dsDNA | 10.0 U/mL | 0.0-15.0 U/mL |
| Anti-MPO | 0.30 U/mL | 0.0-3.0 U/mL |
| Anti-PR3 | 0.30 U/mL | 0.0-3.0 U/mL |
| Cryoglobulins | Negative | Negative |
| C3 | 130.0 mg/dL | 83.0-177.0 mg/dL |
| C4 | 17.3 mg/dL | 12.0-36.0 mg/dL |
| IgA | 195.0 mg/dL | 40.0-350.0 mg/dL |
| IgG | 1075.0 mg/dL | 650.0-1600.0 mg/dL |
| IgM | 130.0 mg/dL | 50.0-300.0 mg/dL |
| Serum protein electrophoresis | ||
| Albumin | 4.05 g/dL | 4.02-4.76 g/dL |
| Alpha-1 globulin | 0.44 g/dL | 0.21-0.35 g/dL |
| Alpha-2 globulin | 0.96 g/dL | 0.51-0.85 g/dL |
| Beta globulin | 0.81 g/dL | 0.57-0.99 g/dL |
| Gamma globulin | 1.00 g/dL | 0.80-1.35 g/dL |
| Total protein | 7.20 g/dL | 6.60-8.70 g/dL |
| Urinalysis | ||
| pH | 5.5 | 4.5-8.0 |
| Protein | Negative | Negative |
| Glucose | Negative | Negative |
| Ketones | Negative | Negative |
| Bilirubin | Negative | Negative |
| Nitrite | Negative | Negative |
| White blood cells | Not detected | ≤11.8/µL |
| Erythrocytes | Not detected | ≤10.1/µL |
| Hyaline casts | Not detected | Usually none |
| Crystals | Not detected | Usually none |
| Protein/creatinine | 0.066 mg/mg | <0.15 mg/mg |
| Coagulation studies | ||
| PT | 13.8 seconds | 11.7-15.3 seconds |
| INR | 1.06 | 0.8-1.2 |
| aPTT | 31.0 seconds | 25.0-34.0 seconds |
| Fibrinogen | 3.1 g/L | 2.0-4.0 g/L |
| Factor V Leiden gene mutation | Not detected | Negative |
| Prothrombin G20210A gene mutation | Not detected | Negative |
| MTHFR gene mutation | Not detected | Negative |
| Protein C activity | 99% | 70.0-140.0% |
| Protein S activity | 56% | 54.7-123.7% |
| Antithrombin activity | 111% | 83.0-128.0% |
| LA | Positive | Negative |
| Anti-β2GPI IgG | 74.0 U/mL | 0.0-10.0 U/mL |
| Anti-β2GPI IgM | 8.2 U/mL | 0.0-10.0 U/mL |
| aCL IgG | 25.0 U/mL | 0.0-40.0 U/mL |
| aCL IgM | 6.5 U/mL | 0.0-40.0 U/mL |
| Investigation | Result | Reference range/interpretation | |
| Pre-treatment | Post-treatment | ||
| Haemoglobin | 9.4 g/dL | 12.9 g/dL | 12.0-15.0 g/dL |
| White blood cells | 1.31 × 109/L | 4.84 × 109/L | 4.50-11.50 × 109/L |
| Neutrophils | 0.1 × 109/L | 2.3 × 109/L | 2.0-7.5 × 109/L |
| Platelet count | 197.0 × 109/L | 154.0 × 109/L | 150.0-450.0 × 109/L |
| ESR | 140.0 mm/h | 28.0 mm/h | 0.0-20.0 mm/h |
| CRP | 8.59 mg/dL | 0.74 mg/dL | < 0.50 mg/dL |
| Procalcitonin | 0.10 ng/mL | 0.04 ng/mL | 0.0-0.50 ng/mL |
| Serum protein | 5.6 g/dL | 6.7 g/dL | 6.6-8.7 g/dL |
| Serum LDH | 176.9 IU/L | 243.0 IU/L | 120.0-246.0 IU/L |
| Serum albumin | 3.8 g/dL | 4.2 g/dL | 3.5-5.0 g/dL |
| Glucose | 82 mg/dL | 88 mg/dL | 74.0-106.0 mg/dL |
| GGT | 10.5 IU/L | 15.8 IU/L | 0.0-38.0 IU/L |
| ALP | 59.0 IU/L | 62.0 IU/L | 25.0-100.0 IU/L |
| AST | 16.0 IU/L | 18.0 IU/L | 3.0-31.0 IU/L |
| ALT | 10.0 IU/L | 13.0 IU/L | 3.0-31.0 IU/L |
| Total bilirubin | 0.7 mg/dL | 0.7 mg/dL | 0.3-1.2 mg/dL |
| Urea | 15.0 mg/dL | 24.0 mg/dL | 19.0-49.0 mg/dL |
| Creatinine | 0.5 mg/dL | 0.6 mg/dL | 0.5 - 1.2 mg/dL |
| SARS-CoV-2 PCR | Positive | Negative | |
| CMV IgG | 137.00 U/mL | ≤12.00 U/mL | |
| CMV IgM | 10.90 U/mL | ≤18.00 U/mL | |
| Parvovirus B19 IgG | <0.1 Index | <0.9 Index | |
| Parvovirus B19 IgM | <0.1 Index | <0.9 Index | |
| EBV VCA IgG | 9.0 U/mL | <18.0 U/mL | |
| EBV VCA IgM | 5.0 U/mL | <36.0 U/mL | |
| HIV 1/2 Ag/Ab combo | Non-reactive | Non-reactive | |
| Aspergillus galactomannan Ag | <0.2 Index | <0.5 Index | |
| Blood culture | Negative | No growth | |
| Urine culture | Negative | No growth | |
| Thoracocentesis (first procedure) | |||
| Fluid appearance | Yellow-orange | Clear or pale yellow | |
| pH | 7.43 | 7.35-7.45 | |
| Protein | 4.1 g/dL | Exudate (pleural fluid protein > 0.5 × serum protein) | |
| LDH | 361.0 IU/L | Exudate (pleural LDH > 0.6 × serum LDH and > 2/3 ULN) | |
| Albumin | 2.5 g/dL | Serum-effusion albumin gradient = 1.3 g/dL | |
| ADA | 32.5 U/L | 0.0-40.0 U/L | |
| Glucose | 89.0 mg/dL | 60-100 mg/dL | |
| Nucleated cells | 776.0/µL | <1000/µL | |
| Differential (neutrophils/lymphocytes) | 71.2/28.8 | Suggests acute inflammation | |
| Pleural fluid culture | Negative | No growth | |
| Thoracocentesis (second procedure) | |||
| Fluid appearance | Yellow-orange | Clear or pale yellow | |
| pH | 7.43 | 7.35-7.45 | |
| Protein | 4.5 g/dL | Exudate (pleural fluid protein > 0.5 × serum protein) | |
| LDH | 332.0 IU/L | Exudate (pleural LDH > 0.6 × serum LDH and > 2/3 ULN) | |
| Albumin | 2.9 g/dL | Serum-effusion albumin gradient = 0.9 g/dL | |
| ADA | 38.4 U/L | 0.0-40.0 U/L | |
| Glucose | 68.0 mg/dL | 60-100 mg/dL | |
| Nucleated cells | 1934.0/µL | <1000/µL | |
| Differential (neutrophils/lymphocytes) | 22.1/77.9 | Suggests a chronic or subacute process | |
| Pleural fluid culture | Negative | No growth | |
| Bone marrow aspiration | |||
| Cellularity | Normocellular aspirate with abundant fragments | Marrow cellularity appropriate for age | |
| Neutrophils | 24% of total cells decreased to terminally mature forms | 40-60% Mild maturation abnormality with arrest at the myelocyte stage | |
| Monocytes | 7% of total cells are in normal maturation | 2-10% Normal | |
| Erythroid cells | 58% of total cells, hyperplasia with occasional nuclear bridges and basophilic stippling | 20-30% Erythroid hyperplasia; reactive stress/accelerated erythropoiesis | |
| Megakaryocytes | Increased cellularity, multiple maturation stages, some giant forms, no dysplastic features | Abnormal reactive megakaryopoiesis, no evidence of dysplasia or myeloproliferative disorder | |
| Lymphocytes | 8.4% of total cells: T, B, NK cells with normal phenotype | 10-20% normal | |
| Plasma cells | 0.02% of total cells Normal phenotype | 0-5% normal | |
| Blasts | 3.7% of total cells, 9% B-lymphoid, 15% immature, 48% granulocytic/neutrophilic, 20% erythroid, 13% monocytic/dendritic lymphoplasmacytoid | ≤5% Heterogeneous blast population, below threshold for acute leukemia | |
| Fibrosis | None | None | |
| Iron stores | Adequate | Normal | |
| Overall impression | Normocellular marrow with discrete maturational abnormalities across hematopoietic lineages | Findings consistent with a reactive process; no evidence of dysplasia, clonal proliferation, or hematological malignancy | |
| Investigation | Result | Reference range |
| LA | Positive | Negative |
| Anti-β2GPI IgG | 59.0 U/mL | 0.0-10.0 U/mL |
| Anti-β2GPI IgM | 9.0 U/mL | 0.0-10.0 U/mL |
| aCL IgG | 19.0 U/mL | 0.0-40.0 U/mL |
| aCL IgM | 7.4 U/mL | 0.0-40.0 U/mL |
| Authors | Age and gender | ssSSc clinical manifestations and diagnostic findings | APS clinical manifestations | Autoantibodies | Temporal relationship | Treatment | Outcome |
| Leite and de Carvalho [ | 48, F | RP, facial telangiectasia, arthralgia, gastroesophageal reflux; scleroderma pattern on nailfold capillaroscopy; subtle centrilobular ground-glass opacities bilaterally; diffuse oesophageal ectasia | Bilateral central retinal vein thrombosis | ANA 1:320, centromere pattern; ACA; aCL IgG; LA | APS manifestations and diagnosis preceded ssSSc features | Enalapril, omeprazole, bromopride, warfarin | No relapse or complications observed at five-year follow-up |
| Rossi et al. [ | 34, M | RP, facial telangiectasia, arthralgia, dyspepsia, dysphagia, asthenia; scleroderma pattern on nailfold capillaroscopy; reduced oesophageal peristaltic pressure, lower oesophageal sphincter dysfunction, grade 2 oesophagitis | Recurrent deep venous thrombosis | ANA 1:640, centromere pattern; ACA; anti-β2GPI IgM | APS manifestations preceded ssSSc features; APS was formally diagnosed after ssSSc | Nifedipine, proton pump inhibitors (unspecified), warfarin | No relapse or complications observed at two-year follow-up |
| Alghamdi and Derbes [ | 59, F | Gastroesophageal reflux disease, dysphagia, persistent gastrointestinal bleeding; gastric and duodenal angioectasias | Recurrent arterial thrombosis of both legs | ANA 1:640, centromere pattern; ACA; anti-RNA polymerase III; aCL IgG; anti-β2GPI IgG; LA | APS manifestations and diagnosis preceded ssSSc features | Argon plasma coagulation, octreotide, hydroxychloroquine, antiplatelet therapy (unspecified) | No relapse or complications observed at two-year follow-up |
| Our case | 20, F | RP, multiple DU, telangiectasias, inflammatory arthralgia, dyspepsia, epigastric pain; scleroderma pattern on nailfold capillaroscopy; mild chronic gastritis | Pulmonary embolism with lingula infarction | ANA 1:2560, centromere pattern; ACA; anti-Ro52; anti-β2GPI IgG; LA | APS diagnosed after establishing ssSSc | Nifedipine, pentoxifylline, intravenous iloprost, sildenafil, bosentan, acetylsalicylic acid, heparin, followed by warfarin | DU healed in two months; four minor isolated uncomplicated DU over 20 months of follow-up; no further thrombosis or organ involvement |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSystemic Sclerosis and Related Diseases · Systemic Lupus Erythematosus Research · Inflammatory Myopathies and Dermatomyositis
Introduction
Systemic sclerosis (SSc) is a rare connective tissue disease characterized by the association of autoimmune features, vasculopathy, and fibrosis of the skin and/or internal organs [1]. Vasculopathy is central to the pathogenesis of SSc, involving complex interactions among endothelial cells, vascular smooth muscle cells, extracellular matrix, and circulating mediators, ultimately leading to vascular remodelling, vasospasm, and vessel occlusion [2]. Raynaud’s phenomenon (RP) and digital ulcers (DUs) are manifestations of this underlying vasculopathy, with critical digital ischemia being the most feared complication [3].
Systemic sclerosis sine scleroderma (ssSSc), first described by Rodnan and Fennell [4], is characterized by the presence of SSc-associated visceral manifestations and serologic abnormalities, in the absence of skin fibrosis [5]. This subset accounts for 8.8% of all patients registered in the international EUSTAR database, where a large cohort of 350 ssSSc patients reported current or previous DU in 4.7% and 19.0% of cases, respectively, and demonstrated a predominance of anti-centromere antibodies (ACA; 61%) over antitopoisomerase antibodies (15.1%) [6]. According to the database, ssSSc patients with DU tended to be younger, had more frequent oesophageal involvement, a higher prevalence of anti-U1RNP antibodies, and elevated creatine kinase levels compared to those without DU [6]. In the presence of DU, it is important to consider the potential coexistence of other causes of vasculopathy, such as large vessel disease, embolism, inflammation, infection, and paraneoplastic syndromes, particularly in cases refractory to standard treatment [7].
Antiphospholipid syndrome (APS) is a systemic autoimmune disease characterized by arterial, venous, or microvascular thrombosis, pregnancy morbidity, or other non-thrombotic manifestations in patients with persistent antiphospholipid antibodies (aPL) [8]. APS can be classified as primary, occurring in isolation, or secondary, when associated with an underlying condition such as a connective tissue disease, malignancy, or medication exposure [9].
According to the Euro-phospholipid Project, SSc accounts for only 0.7% of APS cases [10]. The prevalence of APS among SSc patients is estimated to be approximately 4%, based on a French multicentre prospective study [11]. In patients with ssSSc, secondary APS has been rarely reported [12-14]. To the best of our knowledge, this is the first reported case of ssSSc presenting with recurrent DU and pulmonary embolism attributed to secondary APS.
Case presentation
A 20-year-old Caucasian female was diagnosed with ssSSc at the age of 18, presenting at diagnosis with RP, multiple DU, telangiectasias, inflammatory arthralgias, dyspepsia, and epigastric pain. She denied alcohol, tobacco, or illicit drug use. On physical examination, the patient had no puffy fingers, skin thickening, or calcinosis. Laboratory findings are summarized in Table 1.
Nailfold capillaroscopy showed an early scleroderma pattern, and upper gastrointestinal endoscopy identified mild chronic gastritis. There was no evidence of cardiac or pulmonary involvement.
Initial management included nifedipine 30 mg/day, pentoxifylline 800 mg/day, and intravenous iloprost via an elastomeric pump, yielding partial symptom control. Due to persistent DU, the nifedipine dose was increased to 60 mg/day, and sildenafil (up to 75 mg/day), bosentan (up to 250 mg/day), and acetylsalicylic acid (100 mg/day) were added. Despite optimized therapy, DU continued to recur, requiring ongoing iloprost infusions.
The patient later presented with deep, painful DU on the pulp of the second and third fingers of the left hand, with signs of infection but with preserved distal pulses (Figure 1).
Digital ulcers on the second and third fingers of the left handThe image shows two digital ulcers in a patient with systemic sclerosis sine scleroderma, which were refractory to optimized vasodilator therapy, including calcium channel blockers, phosphodiesterase inhibitors, and regular iloprost infusions, as well as additional strategies such as endothelin receptor antagonists and acetylsalicylic acid.
She was admitted for intravenous iloprost, antibiotics (initially trimethoprim/sulfamethoxazole, later switched to ciprofloxacin), wound care, and pain management, which required opioid analgesia. Further laboratory investigations were conducted, and the results are presented in Table 2.
Colour Doppler ultrasonography showed inflammatory parietal thickening and stenosis of the left radial artery. Mycophenolate mofetil was introduced and titrated to 2 grams/day, based on the suspicion of an underlying inflammatory vasculopathy contributing to ischemia.
Approximately one month later, the patient developed pleuritic chest pain, dyspnoea, and fever, testing positive for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Computed tomography pulmonary angiogram revealed pulmonary thromboembolism, complicated by lingula infarction, and left-sided pleural effusion, necessitating a second hospital admission. Anticoagulation was initiated with low molecular weight heparin (1 mg/kg twice daily), later transitioned to warfarin (target international normalised ratio (INR) of 2-3), and acetylsalicylic acid (100 mg/day) was maintained. Mycophenolate was discontinued, and nirmatrelvir/ritonavir, along with ceftriaxone, were started for presumed bacterial co-infection.
Thoracocentesis of the left-sided pleural effusion yielded exudative fluid; infectious serologies and cultures (blood, urine, pleural fluid) were negative. Despite treatment, the patient developed febrile neutropenia, requiring sequential antibiotic escalation (piperacillin/tazobactam, meropenem, and vancomycin). Bone marrow aspiration confirmed a reactive process, and recovery was achieved with filgrastim. Subsequently, she developed a right pleural effusion and a new pulmonary infarction. A second thoracocentesis revealed sterile exudate. Laboratory test results from this second admission are summarised in Table 3.
After 23 days of broad-spectrum antibiotics, persistent fever, negative cultures, and unremarkable imaging (including positron emission tomography/computed tomography), a non-infectious inflammatory process was assumed. Prednisolone at a dose of 0.5 mg/kg/day was initiated and gradually tapered, leading to sustained clinical and laboratory improvement. Following two months of anticoagulation, the DU healed completely (Figure 2).
Complete healing of the digital ulcers following two months of anticoagulation therapyFollowing the diagnosis of secondary antiphospholipid syndrome, anticoagulation was initiated, first with low molecular weight heparin and later transitioned to warfarin. The image shows complete healing of the digital ulcers on the second and third fingers of the left hand two months after the initiation of this therapy.
Over 24 months of follow-up, the patient experienced four minor, isolated, and uncomplicated DU episodes, likely related to difficulty in maintaining the target INR; these resolved quickly with local wound care. No additional thrombotic events, bleeding events, or new organ involvement were observed. Repeat testing confirmed persistent positivity for lupus anticoagulant (LA) and anti-β2-glycoprotein I (anti-β2GPI) IgG antibodies (Table 4), measured more than 12 weeks apart, establishing the diagnosis of secondary APS.
A timeline summarizing the patient’s key clinical events is shown in Figure 3.
Timeline of major clinical eventsThe figure summarizes the patient’s clinical course from diagnosis in August 2021 through August 2025, highlighting the timing of diagnoses, hospitalizations, thrombotic events, therapeutic interventions, and outcomes, including complete digital ulcer healing and long-term follow-up without new organ involvement.APS: antiphospholipid syndrome; DU: digital ulcers; kg: kilogram; mg: milligrams; ssSSc: systemic sclerosis sine scleroderma
Discussion
We describe a case of ssSSc associated with secondary APS, which, to our knowledge, is the fourth reported case of this clinical association, highlighting its rarity and clinical relevance. Notably, this is the first case to manifest both severe digital ulceration and pulmonary embolism.
The previously reported cases are summarized below. Key clinical characteristics of all cases, including ours, are presented in Table 5.
The first published case report [12] describes a 48-year-old female patient who initially presented with bilateral central retinal vein thrombosis and was subsequently diagnosed with APS after two positive measurements of anticardiolipin antibody (aCL) IgG and LA more than 12 weeks apart. Treatment with warfarin was initiated. Remarkably, her initial immunological workup revealed positive antinuclear antibody (ANA) with a 1:320 centromere pattern and positivity for ACA. Approximately one year later, the patient developed RP, facial telangiectasia, acute polyarthralgia, and gastroesophageal reflux. Nailfold capillaroscopy demonstrated a scleroderma pattern. A chest CT scan showed subtle centrilobular ground-glass opacities bilaterally and diffuse oesophageal ectasia, which was confirmed on oesophagography. Based on these findings, a diagnosis of ssSSc was made, and the patient was started on treatment with enalapril, omeprazole, and bromopride, resulting in clinical improvement and no further complications during a five-year follow-up period.
The second published case report [13] describes a 34-year-old male patient with a history of recurrent deep venous thrombosis. He developed RP, facial telangiectasia, arthralgia, dyspepsia, dysphagia, and asthenia. Laboratory investigation revealed a positive ANA with a 1:640 centromere pattern, positive ACA, and anti-β2GPI IgM antibodies. Nailfold capillaroscopy showed a scleroderma pattern. Oesophagogastroduodenoscopy revealed lower oesophageal sphincter dysfunction with grade 2 oesophagitis, and a high-resolution oesophageal manometry confirmed a reduction in peristaltic pressures. Based on these findings, a diagnosis of ssSSc was established. The patient started nifedipine for RP and a proton pump inhibitor, which provided relief for the gastroesophageal reflux symptoms. The recurrent deep vein thrombosis and the positive anti-β2GPI on two occasions more than 12 weeks apart led to the diagnosis of APS, and the patient was started on warfarin. Over the two-year follow-up period, the patient did not experience any additional complications.
The third published case report [14] describes a 59-year-old African American female with a history of tobacco use, hepatitis C infection, mitral valve stenosis, and recurrent arterial thrombosis of both legs, for which she underwent stenting and was placed on dual antiplatelet therapy. Laboratory investigation revealed high triple-positive aPL on two occasions more than 12 weeks apart, leading to a diagnosis of APS. Four years later, the patient developed mild symptoms of gastroesophageal reflux disease and dysphagia, along with persistent gastrointestinal bleeding. Endoscopic studies revealed gastric and duodenal angioectasias, which were treated with argon plasma coagulation and octreotide therapy. Laboratory assessment showed a positive ANA with a 1:640 centromere pattern and positive ACA and anti-RNA polymerase III antibodies. The diagnosis of ssSSc was assumed, and the patient was started on hydroxychloroquine 400 mg/day. Given the high risk of bleeding, antiplatelet therapy was maintained over anticoagulation for APS management. Over a two-year follow-up period, no further thrombotic episodes or SSc-related symptoms were observed.
In our case, recurrent DU represented the main clinical challenge, proving refractory to standard therapies. These included optimized oral vasodilators (a slow-release calcium-channel blocker, a phosphodiesterase type 5 inhibitor, and a prostacyclin analogue), adjuvant antiplatelet therapy, and an endothelin receptor antagonist as a preventive measure to reduce the incidence of new DU. However, following the thrombotic event and initiation of anticoagulation therapy, the ulcers showed a significant improvement, with complete healing and only mild, isolated recurrences over a 20-month follow-up, possibly related to difficulties in maintaining stable anticoagulation levels. Retrospectively, it can be postulated that the underlying APS may have contributed to the pathogenesis of the ulcers, potentially explaining their refractoriness to standard treatment.
The pulmonary embolism was likely multifactorial, with SARS-CoV-2 infection acting as a possible trigger due to its recognized prothrombotic potential - although this effect is typically more pronounced in severe cases requiring intensive care and cardiorespiratory support [15]. The presence of underlying APS, together with SSc-associated vasculopathy, likely potentiated the risk of pulmonary embolism.
Compared with previously reported cases, our patient presented at a younger age, consistent with EUSTAR data showing that ssSSc patients with DU tend to be younger [6]. ACA positivity in all cases and oesophageal involvement in the three earlier reports - defined by clinical symptoms and/or complementary investigations - also align with the referred registry [6].
In our case, ssSSc features were evident prior to the thrombotic events, leading to the diagnosis of ssSSc before APS was identified. In contrast, in the earlier case reports, APS manifestations preceded the development of SSc features, although APS was formally diagnosed after ssSSc in the second case [13].
No associations between SSc-related clinical manifestations and APS have been reported, apart from a higher frequency of anti-topoisomerase I (anti-Scl-70) positivity in SSc patients with secondary APS compared to those with isolated SSc [11].
Several studies have found statistically significant associations between aPL positivity and various SSc clinical features - some have reported a link between aPL and the development of DU [11,16], with anti-β2GPI identified as an independent risk factor for active DU in both univariate and multivariate analysis [11]; one study reported a link with interstitial lung disease, where elevated aCL IgM and IgG levels were associated with increased risk [16]; the same study found an association between RP and positive aCL IgM [16]; higher aCL titres were associated with an increased risk of venous thrombosis [17]. In contrast, other studies have failed to demonstrate significant associations between aPL positivity and SSc-related clinical features [18,19].
However, it is important to note that most of these studies involved relatively small patient samples, which can result in insufficient statistical power and contribute to the heterogeneous findings, thereby precluding firm conclusions. Moreover, in most of these studies, aPL measurements were not repeated after the recommended 12-week interval, as required to confirm the diagnosis of APS. Furthermore, the aPL titres observed in these studies have often been below the diagnostic thresholds for APS.
Based on the discussed data and its limitations, the current evidence does not justify routine screening for aPL in SSc. Exceptions may include situations such as pregnancy planning, recurrent thromboembolic events, or DU refractory to optimized therapy, where an underlying APS may play a contributory role [9,20].
This case report contributes to the literature by documenting a rare presentation of ssSSc associated with secondary APS, complicated by DU and pulmonary embolism. It highlights the importance of early recognition and integrated management, providing clinicians with practical insights into diagnostic strategies and therapeutic decisions.
Conclusions
SSc is a complex disease, with DU representing one of its most challenging and debilitating manifestations. In cases of recurrent, treatment-refractory DU, the association with APS should be suspected, and appropriate screening should be performed, particularly if additional thrombotic features are present, as early identification and targeted therapy may significantly improve patient outcomes.
While the association between SSc and APS remains incompletely understood, further research is essential to clarify the underlying mechanisms, their impact on disease presentation and progression, and the most effective treatment strategies. A better understanding of this association could lead to improved diagnostic accuracy and enable more targeted therapeutic approaches.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Systemic sclerosis Lancet Denton CP Khanna D 1685169939020172841306410.1016/S 0140-6736(17)30933-9 · doi ↗ · pubmed ↗
- 2Vascular involvement in systemic sclerosis (scleroderma)J Inflamm Res Pattanaik D Brown M Postlethwaite AE 105125420112209637410.2147/JIR.S 18145 PMC 3218751 · doi ↗ · pubmed ↗
- 3Consensus best practice pathway of the UK Scleroderma Study Group: digital vasculopathy in systemic sclerosis Rheumatology (Oxford) Hughes M Ong VH Anderson ME 201520245420152611615610.1093/rheumatology/kev 201 · doi ↗ · pubmed ↗
- 4Progressive systemic sclerosis sine scleroderma JAMA Rodnan GP Fennell RH Jr 66567018019621449314210.1001/jama.1962.03050210027006 · doi ↗ · pubmed ↗
- 5Systemic sclerosis sine scleroderma Adv Clin Exp Med Kucharz EJ Kopeć-Mędrek M 8758802620172906858610.17219/acem/64334 · doi ↗ · pubmed ↗
- 6Cutaneous manifestations, clinical characteristics, and prognosis of patients with systemic sclerosis sine scleroderma: data from the International EUSTAR database JAMA Dermatol Lescoat A Huang S Carreira PE 83784715920233737899410.1001/jamadermatol.2023.1729 PMC 10308295 · doi ↗ · pubmed ↗
- 7Differential diagnosis of critical digital ischemia in systemic sclerosis: report of five cases and review of the literature Semin Arthritis Rheum Sharp CA Akram Q Hughes M Muir L Herrick AL 2092164620162731862710.1016/j.semarthrit.2016.05.001 · doi ↗ · pubmed ↗
- 82023 ACR/EULAR antiphospholipid syndrome classification criteria Ann Rheum Dis Barbhaiya M Zuily S Naden R 125812708220233764045010.1136/ard-2023-224609 · doi ↗ · pubmed ↗