Identification and Exploration of a Series of SARS-Cov‑2 MPro Cyano-Based Inhibitors Revealing Ortho-Substitution Effects within the P3 Biphenyl Group
Emma Clyde-Allen, Mikołaj Zmudzinski, Mohammad Afsar, Ciyana James, Anindita Nayak, Digant Nayak, Priscila dos Santos Bury, Dirk Jochmans, Johann Neyts, Christopher J. Scott, Shaun K. Olsen, Marcin Drag, Rich Williams

TL;DR
This paper explores the development of a new SARS-CoV-2 protease inhibitor by modifying a compound's structure to improve its effectiveness and selectivity.
Contribution
The study introduces a novel approach using ortho-substituted biphenyl groups to enhance inhibitor potency and binding.
Findings
Compound 6a showed submicromolar potency against SARS-CoV-2 MPro.
Ortho-substitution in the P3 benzamide improved biochemical and cellular activity.
Compound 22e, with a proline modification, achieved high potency and selectivity.
Abstract
Starting from a simple scaffold hopping exercise based on our previous exploration of cysteine protease inhibitors against legumain, compound 6a was identified as a starting point for the development of a SARS-CoV-2 main protease (MPro) inhibitor. Compound 6a displayed submicromolar biochemical potency in the ultrasensitive assay developed by Drag and co-workers. Through an iterative structure–activity relationship campaign, we discovered an unexpected improvement in both biochemical and cellular potency through the incorporation of an ortho substituent within the P3 benzamide. X-ray crystallography revealed that incorporation of the ortho substituent caused a subtle but important binding enhancement of the P1 glutamate group within the MPro S1 pocket. While incorporation of the ortho substituent improved the potency, the off-target selectivity against a panel of cysteine proteases and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1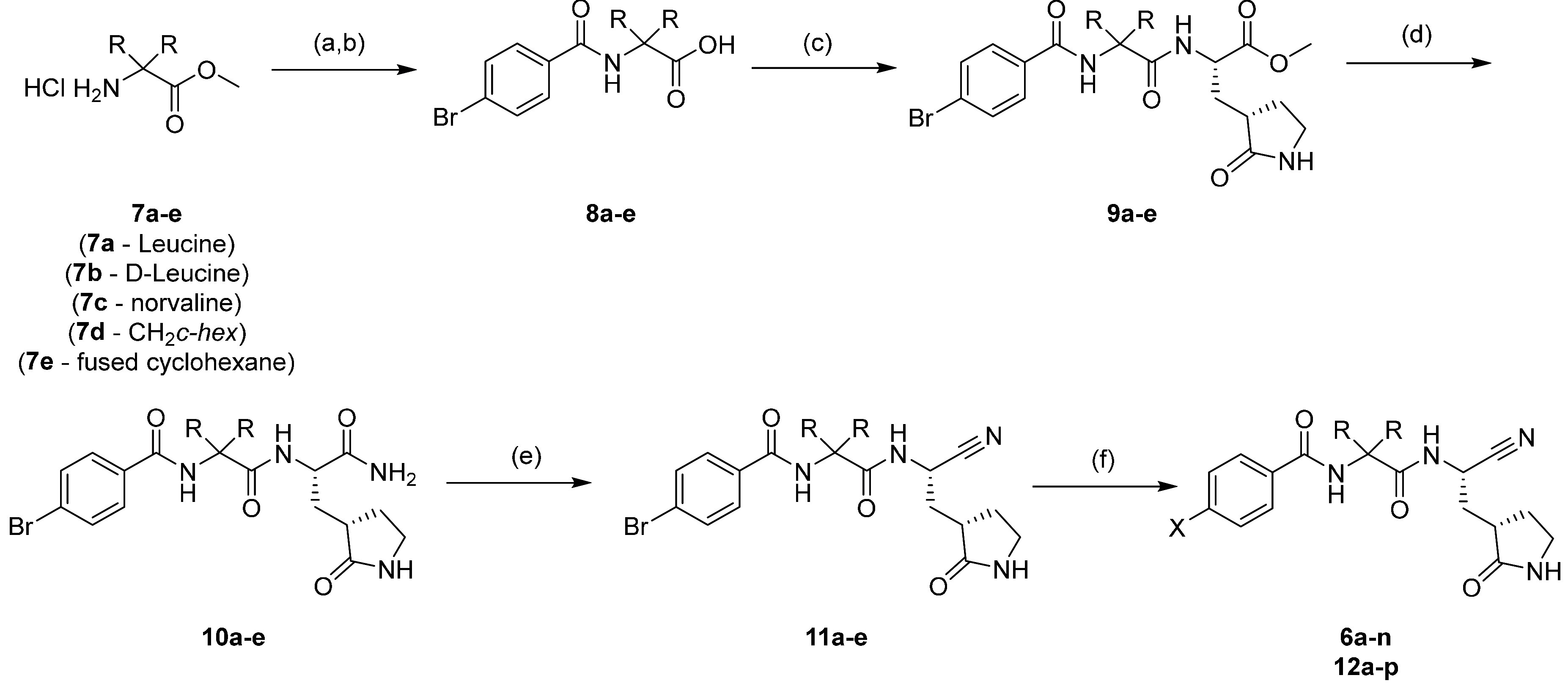 1
1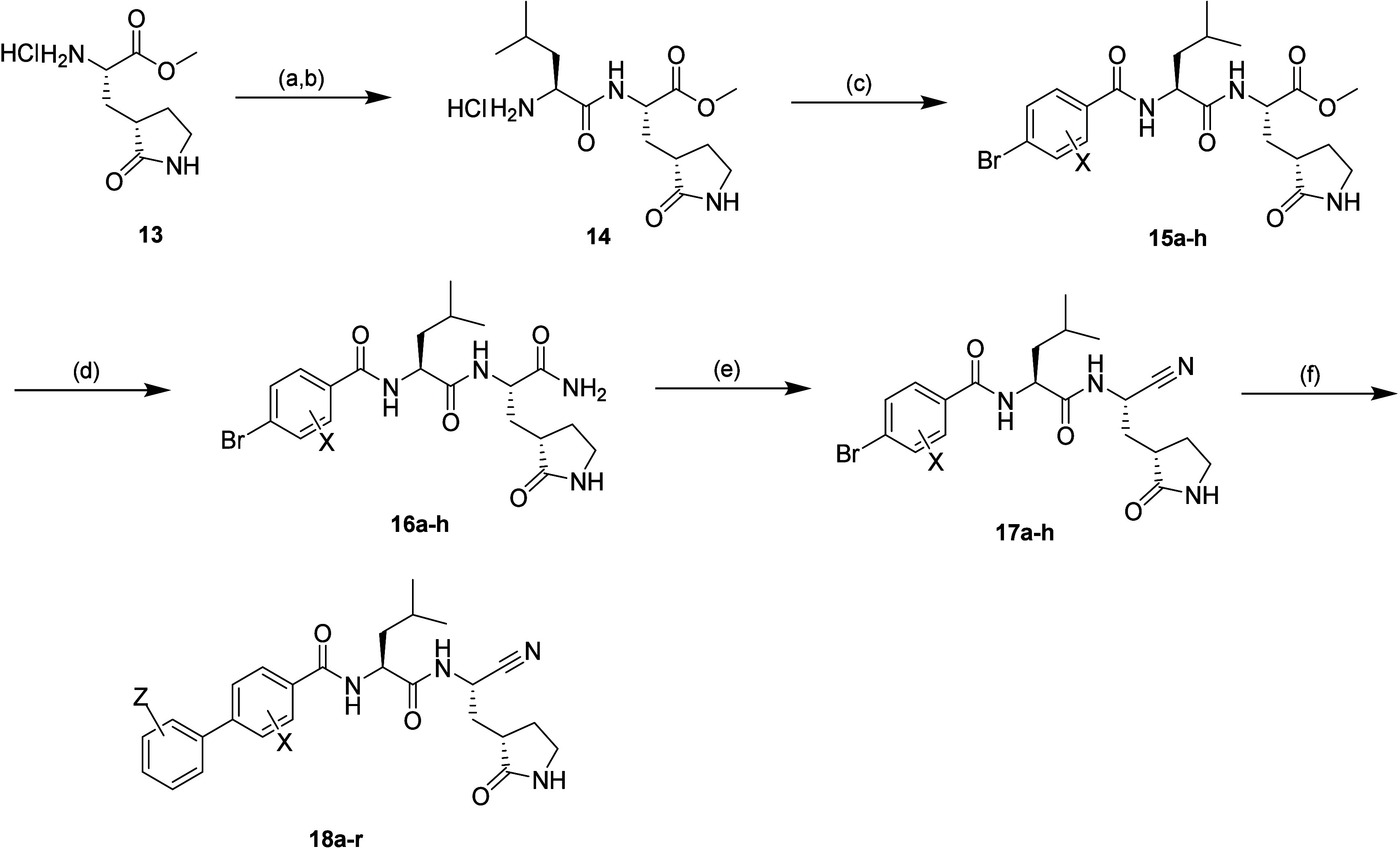 2
2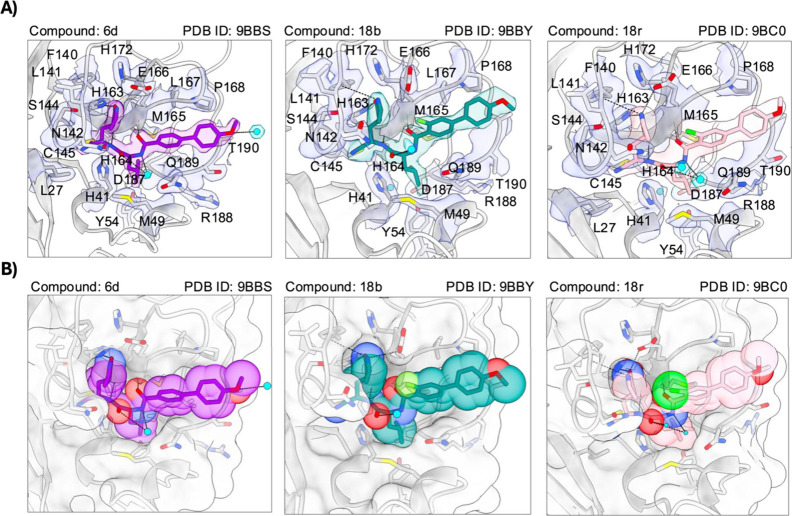 2
2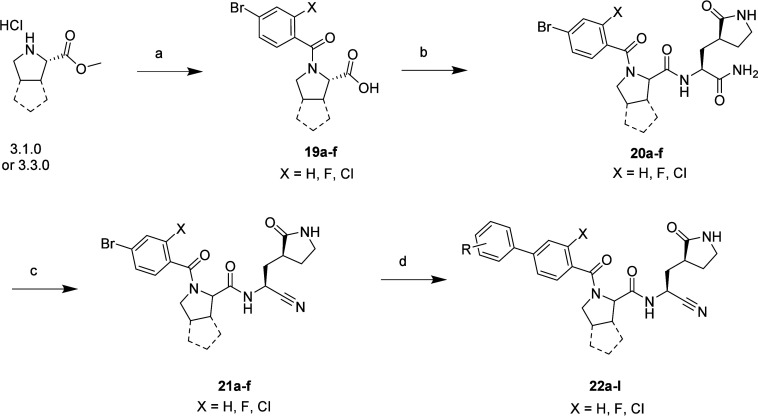 3
3| MPro IC50 (μM) | EC50 (μM) SARS-CoV-2/VeroE6-GFP | CC50 txicity (μM) in cells | |
|---|---|---|---|
|
| 0.786 | 12.5 ± 0.50 | >100 |
|
| 0.859 | 11.9 ± 0.10 | >100 |
|
| 0.622 | 12.1 ± 0.50 | >100 |
|
| 0.560 | 11.1 ± 0.30 | >100 |
| compound | MPro IC50 (nM) | CatS IC50 (nM) | CatK IC50 (nM) | CatB IC50 (nM) | CatL IC50 (μM) |
|---|---|---|---|---|---|
| nirmatrelvir | N/A | 670.0 | 1400 | 1300 | >10 |
|
| 162.7 | 27.9 | 48.5 | 142.3 | >10 |
|
| 178.1 | 232.4 | 77.0 | 106.3 | >10 |
|
| 49.0 | 15.5 | 328.9 | 114.6 | >10 |
|
| 45.0 | 24.6 | 123.5 | 193.4 | >10 |
| IC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|
| compound | R | X | MPro | CatS | CatK | CatB | CatL |
| nirmatrelvir | 670.0 | 1400 | 1300 | >10 | |||
|
| – | Cl | 0.049 | 0.015 | 0.329 | 0.115 | >10 |
|
| 3-MeO-Phe | H | 1.02 | ND | ND | ND | ND |
|
| 4-MeO-Phe | H | 1.13 | ND | ND | ND | ND |
|
| 3-MeO-Phe | F | 0.465 | ND | ND | ND | ND |
|
| 4-MeO-Phe | F | 0.394 | ND | ND | ND | ND |
|
| 4-MeO-Phe | Cl | 0.087 | 1.03 | 2.08 | >10 | >10 |
|
| 3-MeO-Phe | Cl | 0.120 | 1.29 | 4.52 | >10 | >10 |
|
| 3-MeO-Phe | H | 2.67 | ND | ND | ND | ND |
|
| 4-MeO-Phe | H | 2.99 | ND | ND | ND | ND |
|
| 3-MeO-Phe | F | 1.91 | ND | ND | ND | ND |
|
| 4-MeO-Phe | F | 1.27 | ND | ND | ND | ND |
|
| 4-MeO-Phe | Cl | 0.743 | 0.718 | 0.680 | >10 | >10 |
|
| 3-MeO-Phe | Cl | 1.06 | 0.538 | 1.07 | >10 | >10 |
|
| 4-MeO-Phe | H | 1.76 | 0.011 | >10 | >10 | >10 |
|
| 3-MeO-Phe | H | 2.01 | 0.006 | 1.64 | >10 | >10 |
|
| 4-MeO-Phe | F | 0.807 | 0.016 | >10 | >10 | >10 |
|
| 3-MeO-Phe | F | 0.691 | 0.059 | 1.34 | 0.238 | >10 |
|
| 4-MeO-Phe | Cl | 0.077 | 0.011 | 1.52 | 0.104 | >10 |
|
| 3-MeO-Phe | Cl | 0.094 | 0.011 | 0.887 | 0.137 | >10 |
| MPro IC50 (μM) | EC50 (μM) SARS-CoV-2/VeroE6-GFP | CC50 toxicity (μM)
in cells |
| LogD (pH 7.4) | mouse, human
microsomal stability (Clint) (μL min–1 mg–1) | |
|---|---|---|---|---|---|---|
| nirmatrelvir | 0.101 ± 0.2 | >100 | – | – | – | |
|
| 0.049 | 2.18 ± 0.2 | >100 | 34 | 3.1 | 111, 40 |
|
| 0.087 | 0.289 ± 0.05 | >100 | 97 | 3.4 | 450, 183 |
|
| 0.120 | 0.331 ± 0.05 | >100 | 82 | 3.5 | 450, 209 |
|
| 0.077 | 2.49 ± 0.20 | >100 | – | – | – |
|
| 0.095 | 8.64 ± 0.30 | >100 | – | – | – |
- —National Institutes of Health10.13039/100000002
- —National Institutes of Health10.13039/100000002
- —National Institute of General Medical Sciences10.13039/100000057
- —National Institute of General Medical Sciences10.13039/100000057
- —National Institute of General Medical Sciences10.13039/100000057
- —Cancer Prevention and Research Institute of Texas10.13039/100004917
- —Cancer Prevention and Research Institute of Texas10.13039/100004917
- —Argonne National Laboratory10.13039/100006224
- —Department of Education, Northern Ireland10.13039/100008304
- —Office of Research Infrastructure Programs10.13039/100016958
- —Department for economy/UKRINA
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsComputational Drug Discovery Methods · Synthesis and biological activity · Multicomponent Synthesis of Heterocycles
The most recent human coronavirus disease, COVID-19, originated in Wuhan City, Hubei Province, Central China, when in early December, a number of cases of pneumonia with unknown etiology occurred. ?−? ? ? Following genome sequencing analysis of samples taken from the lower respiratory tract of some of these patients, the causative agent was identified as a novel coronavirus with >75% sequence homology to SARS-CoV and thus was appropriately named SARS-CoV-2. ?,? The three lethal respiratory hCoVs (SARS-CoV, MERS-CoV, and SARS-Cov-2) tend to infect the lower, rather than the upper, respiratory tract and in severe cases can lead to acute lung injury, acute respiratory distress syndrome (ARDS), septic shock, multiorgan failure, and death. ?−? ? ?
The urgency for therapeutics led scientific research toward drug repurposing strategies to reduce the risk of failure at clinical trial and the time and cost associated with novel drug development. Various anti-inflammatory and anticoagulant drugs have been granted FDA emergency use authorization to be used as a component of a combinational therapy (including baricitinib, dexamethasone, and heparin). However, remdesivir, molnupiravir, and nirmatrelvir are the only direct-acting antivirals approved for emergency use against SARS-CoV-2, of which the latter two are novel SARS-CoV-2 antivirals. Remdesivir was the first to receive this authorization as a broad-spectrum RNA-dependent RNA polymerase (RdRp) intravenous inhibitor. Molnupiravir is a novel β-d-N ^4^-hydroxycytidine-5′-isopropyl ester compound that is taken orally to induce viral RNA mutagenesis and generate nonfunctional viruses.? Nirmatrelvir is a novel covalent oral main protease (M^Pro^) inhibitor taken in combination with ritonavir (a CYP3A4 inhibitor, used in this case as a pharmacokinetic enhancer to overcome poor metabolic stability).? In addition, a series of M^Pro^ mutations have arisen that infer resistance to nirmatrelvir, highlighting the need for additional inhibitors in this field. ?−? ? ?
M^Pro^ is considered an attractive target for development due to its vital role in viral replication. M^Pro^ uniquely recognizes and cleaves following a glutamine residue at the P1 position, which all human cysteine proteases are incapable of doing, reducing the risk for serious toxic side effects. The M^Pro^ active site contains a Cys145–His41 catalytic dyad. Covalent inhibitors utilize a warhead group that will react with Cys145 in the active site to block catalytic activity. ?−? ? ? ? ? Throughout the study, a nitrile warhead was used to allow the inhibitors to bind covalently (to produce tightly bound inhibitors) but reversibly to prevent nonselective binding (as per standard practice in our lab) and sequential toxicity observed with more reactive and irreversible warheads. ?−? ?
Plan of Investigation
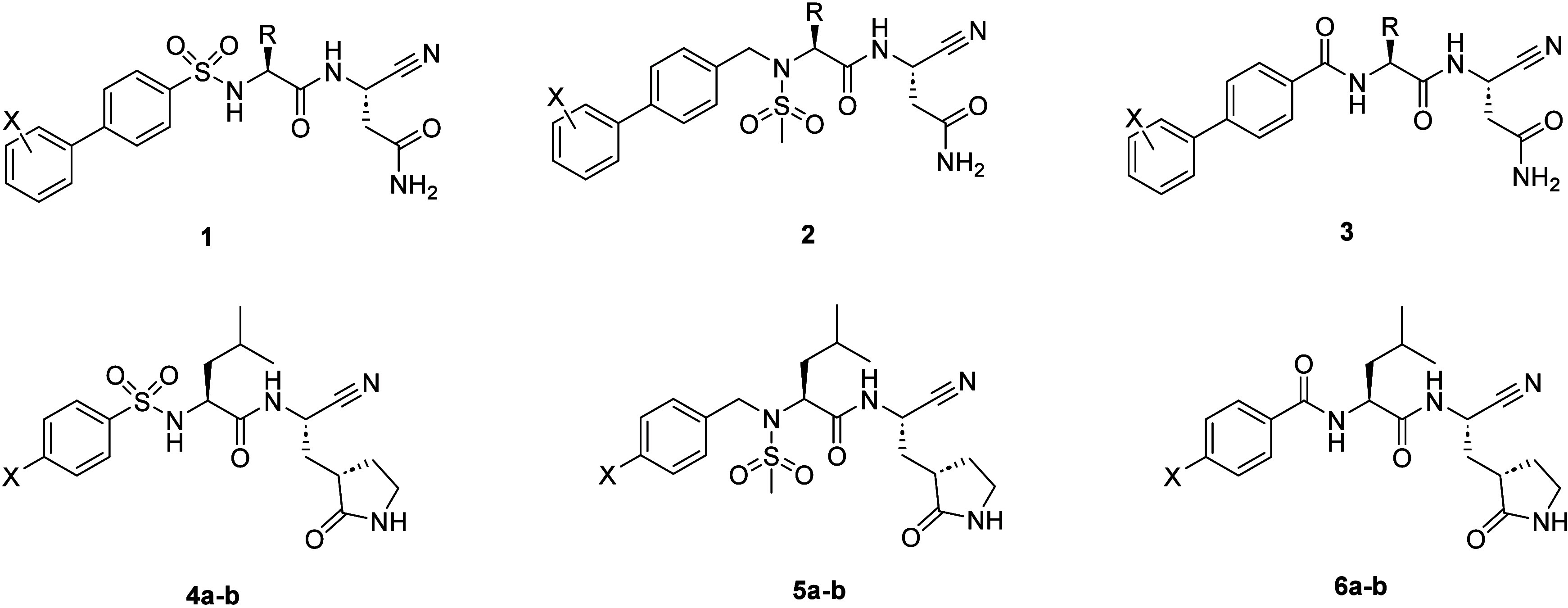
To identify a new inhibitor scaffold to target SARS-CoV-2 M^Pro^ we utilized a scaffold hopping exercise based on a series of inhibitors developed against another cysteine protease target, legumain, from our lab (1–3; Figure). ?−? ? Both proteases have a very distinct requirement for either an Asn (legumain) or Gln (M^Pro^) at the P1 position. ?−? ? In addition, the reported preferences of P2 groups were relatively similar, with small lipophilic groups being optimal for both M^Pro^ and legumain. Compounds 4a–6b were readily synthesized following published procedures utilized for scaffolds 1–3. ?−? ? Screening of these initial test compounds within the assay developed by Drag and co-workers revealed that compounds 4a and 4b and compounds 5a and 5b (tested as both racemates and single diastereomers) were completely inactive against both viral proteases, M^Pro^ and PL^Pro^. The benzamide-scaffold-based compounds 6a and 6b were active below 100 μM and displayed submicromolar biochemical potency (Table). This result, coupled with the lack of activity against the main off target PLpro, warranted further investigation with the aim of identifying an inhibitor with excellent biochemical potency (IC_50_ ≤ 100 nM), subtype selectivity (>100 fold), and cell-based antiviral potency (EC_50_ < 500 nM) in the VeroE6 model.
Different classes of legumain inhibitor scaffolds developed in the lab and, using a scaffold hopping exercise, translated via changing P1 (Asn to cyclic Glu) to afford a test set of MPro inhibitors for screening.
1: Screening Results for 6a–n, 11b–e, and 12a–p
Chemistry
The synthesis of the core bromo analogues (11a–e; Scheme) was conducted from commercially available methyl esters of amino acids. The hydrochloride salts were coupled under standard conditions with HATU in DMF to obtain the corresponding benzoic acids. The subsequent methyl esters from the coupling reaction were then saponified under mild basic conditions to afford the carboxylic acids (8a–e; Scheme). The subsequent carboxylic acids were then coupled to the cyclic glutamate methyl ester in good to excellent yields to install the P1 group (9a–e). Conversion of the methyl ester to the primary amide was carried out using ammonium hydroxide in methanol in a sealed vessel, and the amide in turn was dehydrated with cyanuric chloride to install the cyano warhead in moderate to excellent yield (11a–e). Following an approach similar to that explored in the development of the legumain inhibitors, these core bromo intermediates were used to generate a small library of P3 biaryl analogues (6a–n) using aryl- and heteroarylboronic acids that we have observed to be productive in affording active protease inhibitors (Scheme). The synthesis of compounds 4a, 4b, 5a, and 5b is given in the Supporting Information.
Synthesis of 6a–n and 12a–p
To explore the SAR of the P3 benzamide ring, an additional synthetic route was utilized (Scheme). The cyclic P1 glutamate mimetic 13 was coupled with Boc-Leu-OH under standard amide coupling conditions, and the subsequent compound was deprotected to afford the desired HCl salt 14 in good overall yield. Coupling of 14 with selected benzoic acids using HATU afforded compounds 15a–h in good to excellent yields. Conversion of the P1 methyl ester was carried out using ammonium hydroxide in methanol in a sealed vessel, as with the previous route, to afford 16a–e. To install the cyano warhead, the primary amide was dehydrated with cyanuric chloride to afford 17a–e in excellent yield. The bromo group was once again used as a chemical handle to perform a Suzuki reaction to afford 18a–r (Scheme).
Synthesis of 18a–r
Results
All of the generated analogues were then screened against both viral proteases, M^Pro^ and PL^Pro^, in a single concentration to determine the % inhibition at 100 μM. Only compounds that displayed >95% inhibition were taken forward for IC_50_ determination. From this initial screen, it was observed that most of the synthesized compounds were active against M^Pro^ with no observable activity against PL^Pro^ (reported as <50% activity; Table). For the P2 SAR, we explored the use of small lipophilic amino acids, such as leucine, norvaline, and the non-natural amino acids (Table). Following reported data, the use of leucine in P2 was optimal, with all compounds tested displaying submicromolar potency against M^Pro^, except for 6g, 6h, and 6k, which showed activity slightly over 1 μM. Incorporation of the unnatural isomer of leucine in P2, while tolerated, did lead to a dropoff in activity (IC_50_ = 0.724 μM for 6a vs 3.24 μM for 12a; Table). The same observation was made upon switching to norvaline, which led to a 7–10-fold dropoff in biochemical potency (IC_50_ = 0.735 μM for 6d vs 5.483 μM for 12f; Table). The use of the cyclohexylmethyl group in P2 did restore some of this activity; however, no submicromolar potency was observed with these alternative P2 groups (Table). As with previously reported peptide-based inhibitors, the use of leucine in P2 was found to be optimal within this series of inhibitors. The SAR of the P3 biphenyl group is robust, with the incorporation of halogens, simple ethers, and heterocycles being well-tolerated with biochemical potencies from 560 nM to 1.3 μM (Table).
The most active inhibitors from the P3 screen (6e, 6f, 6j, and 6l) were selected for testing in the cell-based antiviral assay. This assay focuses on testing the ability of a compound to block replication of the SARS-CoV-2 variant in VeroE6 cells. As M^Pro^ plays a vital role in viral replication, this is an ideal cellular model to test M^Pro^ inhibitors. The results from this screen revealed that all four inhibitors were able to block viral replication in the 11–12 μM range (Table), which was encouraging, although they were significantly less active than the Pfizer inhibitor nirmatrelvir (EC_50_ = 100–180 nM). Further improvements in the cell activity were clearly required. Encouraging for this class of inhibitors, none of the tested compounds displayed toxicity at the highest concentration tested (Table).
2: Cell-Based Antiviral Assay and Toxicity Results for 6e, 6f, 6j, and 6l
To assess whether additional improvements in biochemical and, more importantly, cellular activity could be achieved, further exploration of the P3 benzamide group position was explored. The focus was placed on exploring the SAR of the benzamide phenyl ring. Incorporation of fluorine in close proximity to an amide group has been reported to improve intracellular activity in a number of reported small molecules, such as the ROCK kinase inhibitor.? To test whether this theory could improve cellular activity, a series of mono- and difluorinated P3 inhibitors (18a–i; Scheme) were synthesized. In addition, to challenge this hypothesis and to further expand understanding of the SAR, 2-methyl (18j and 18k), 2-CF_3_ (18l and 18m), and 2-Cl (18n–r) analogues were also synthesized. Each of these inhibitors was tested against M^Pro^ for biochemical potency. All of these analogues were found to be comparably or more potent compared with the unsubstituted phenyl compounds previously tested (Table). The o-fluoro analogues (18a–c), were found to provide a 3–4-fold improvement in potency versus the unsubstituted progenitors (IC_50_ = 0.859 μM for 6f vs 0.178 μM for 18b; Tables and ?), with a similar trend being observed with the 2,3-difluoro (18d and 18e) and 2,5-difluoro (18f and 18g) analogues. The m-fluoro analogues did not replicate this finding (IC_50_ = 0.162 μM for 18a vs 0.661 μM for 18i; Table), highlighting the importance of the ortho position to produce this effect. Testing of compounds 18a–c in the viral infection model also revealed an improvement of between 2- and 3-fold (EC_50_ = 4.18–6.34 μM; Table) versus the unsubstituted progenitor compounds 6e, 6f, 6j, and 6l (EC_50_ = 11.1–12.5 μM; Table). Further screening of the synthesized inhibitors revealed that additional improvements in potency could be achieved by replacing the o-fluoro group with either a methyl, CF_3_, or chloro group (18j–r, IC_50_ = 0.045–0.163 μM; Table). These inhibitors also provided an additional improvement in biochemical potency versus both the unsubstituted phenyl (4–12-fold versus 6e–l, IC_50_ = 0.560–1.303 μM; Table) and the 2-fluoro analogues (2–4-fold versus 18a–c, IC_50_ = 0.162–0.302 μM). In addition, the cellular potency was enhanced especially with compounds 18n–r (IC_50_ = 0.045–0.068 μM) by a similar magnitude via the incorporation of the o-chloro group (Table).
3: Cell-Based Antiviral Assay and Toxicity Results for 18a–r
X-ray Crystallography
To further understand the reason behind these observed improvements in biochemical and cellular potency, we cocrystallized a number of compounds with M^Pro^. The use of cocrystal structures has been used to support the further development of several M^Pro^ inhibitors. ?−? ? ? ? ? Lead inhibitors from each iteration (6d, 18b, and 18r in Figure, others shown in the Supporting Information) were selected to illustrate the impact of variations in the ortho position of the benzamide ring. The high-resolution structures show the presence of the electron density for selected compounds within the active site of M^Pro^ highlighted by interactions with key amino residues (FigureA). The active site Cys145 of M^Pro^ forms the expected covalent bond with the nitrile group of all ligands, resulting in the observed thioimidate products (FigureA). Glu166, His172, His163, Phe140, and Ser144 are present in the hydrogen-bonding range with the P1 group of these compounds. The added fluorine at the ortho position of the P3 group in compound 18b projects toward the P1 group, which may decrease the conformational heterogeneity of the P1 group, including where the covalent adduct is formed with M^Pro^ Cys145. In the structure, the o-fluoro group fills the space between the P3 and P1 groups and locks the inhibitor in place more efficiently (FigureA, 18b). Furthermore, the replacement of fluorine with chlorine, Me, and CF_3_, which are bulkier than fluorine, in the P3 group in compounds 18r, 18k, and 18m, respectively, further restricts the movement of the P1 group and enhances the binding within the S1 pocket (FigureA,B, 18r). This ortho-substitution effect is supported by the improvements observed in the biochemical potency.
(A) Co-crystal structures of compounds 6d, 18b, and 18r in complex with the main protease (MPro). The electron density maps of the compounds and their neighboring active-site residues are shown at a σ contour level of 1.0. The catalytic Cys145 forms a covalent bond with the nitrile group of each ligand, leading to the formation of thioimidate products. Key active-site residues, including Glu166, His172, His163, Phe140, and Ser144, interact with the P1 group of the compounds via hydrogen bonding, contributing to ligand stabilization and inhibitory potency. (B) Surface representations of MPro in complex with compounds 6d, 18b, and 18r, illustrating how each inhibitor is positioned within the enzyme’s active site. The visualizations highlight key interactions with residues Cys145, His41, Glu166, His172, His163, Phe140, and Ser144, which are essential for ligand binding and enzymatic inhibition. Additional cocrystal structures and statistics tables for all structures are contained within the Supporting Information.
Selectivity Profiles of the Lead MPro Inhibitors
The lead compounds identified from this study were screened against a panel of known cysteine proteases and benchmarked against the Pfizer M^Pro^ inhibitor nirmatrelvir (Table). The Pfizer compound was found to have reasonable selectivity across the selected panel of proteases tested. Inhibitors 18a–r were all found to be nonselective versus cathepsin S, K, and B, although no activity against cathepsin L was observed, similar to the Pfizer compound. This lack of selectivity against the panel of cysteine proteases was a concern and could potentially lower the effect in vivo. Interestingly, the incorporation of the o-halo group, especially switching from o-fluoro (18a and 18b) to o-chloro (18n and 18r), also led to improvements in biochemical potency against cathepsin S. The same changes had the opposite effect against cathepsin K, with the incorporation of o-chloro leading to a reduction in biochemical potency, while the cathepsin B and L activities remained unaffected (Table).
4: Selectivities Assessed against Human Recombinant Cathepsins K–L in the Presence of a Fluorogenic Substrate (Z-Phe-Arg-AMC for Cat K, L, and B and Z-Val-Val-Arg-AMC for Cat S)
Next, we aimed to improve the selectivity of this novel series of biphenyl benzamide SARS-CoV-2 M^Pro^ inhibitors. The S1 pocket is well-conserved across all cathepsins; however, the S2 pocket is less well-conserved. It was hypothesized that the shallow S2 pockets of cathepsin K and B could be exploited to achieve better selectivity by exploring larger hydrophobic P2 groups. ?,? Inspired by X-ray crystal structures of SARS-CoV-2 M^Pro^ in complex with the hepatitis C antivirals boceprevir and telaprevir, bicycloproline P2 moieties have been incorporated as cyclic leucine mimetics in M^Pro^ inhibitor scaffolds. ?−? ? ? ? Both bicycloproline P2 moieties ((1R,2S,5S)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-formamide and (1S,3aR,6aS)-octahydrocyclopenta[c]pyrrole-1-formamide, respectively) suitably occupy the M^Pro^ S2 pocket.? Nirmatrelvir also features a (1R,2S,5S)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-formamide P2. Nirmatrelvir displayed inhibitory activity against cathepsins S, K, and B; however, it had a better selectivity profile against the selected human cathepsins than compounds with an isobutylleucine P2. While aiming to improve the selectivity of this series of biphenyl benzamide M^Pro^ inhibitors, the (1R,2S,5S)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-formamide and (1S,3aR,6aS)-octahydrocyclopenta[c]pyrrole-1-formamide bicyclic proline P2 groups were explored alongside further SAR exploration of the cyclohexylmethyl P2 group, which had been previously been shown to lead to improvements in cell effects.
To explore the incorporation of alternative P2 groups, such as bicyclic systems that have been reported in other protease inhibitors, a chemical synthetic route similar to that utilized in the synthesis of P2 leucine inhibitors (Scheme and ?) was explored. ?,? The commercially available methyl esters were converted to the subsequent benzamides (19a–f; Scheme). Incorporation of the ortho substituent was also explored with 2-fluoro- and 2-chlorobenzoic acids as well as the unsubstituted benzoic acid to explore whether the same effect on biochemical and cellular activity as with the progenitor leucine-based inhibitors would be observed. Conversion of the primary amide with cyanuric chloride installed the cyano warhead (20a–f) in excellent yield. This was followed by Suzuki coupling to install the optimal biphenyl groups observed previously. As reported by Pfizer, the [3.1.0]-based compounds were afforded as mixtures of rotamers around the P3 amide bond.? A series of attempts were made to separate these rotamers at both the bromo precursors (21a–c) and the final analogues (22a–c) by standard and chiral preparatory methods. While separation was possible, they rapidly re-established a mixture of rotamers in solution. While this observation was disappointing in terms of being able to further interrogate the SAR of each rotamer, we tested each of the inhibitors as mixtures (see the Supporting Information). In addition to the bicyclic P2 variants, the cyclohexylmethyl P2 was also explored in combination with the o-halo biphenyl P3 groups (12l–k and 23a–d).
Synthesis of 19–22
From this study, it was clear that incorporation of the o-halo group in P3 provided a similar improvement in biochemical potency with all three P2 groups (Table). In each series, moving from the unsubstituted to 2-F and then 2-Cl afforded an improvement, although this was less pronounced with the [3.3.0] P2 group (22g–l; Table). In addition, the use of this bicyclic P2 failed to offer any inhibitors that matched 18n in terms of activity against M^Pro^. Comparable activity to 18n was observed with compounds 18d, 18e, 22e, and 22f, which all displayed excellent biochemical potency (Tables and ?). All four of these inhibitors contained the o-chloro group. Next our attention turned to selectivity profiling of the most active inhibitors from this study. Use of the [3.1.0] bicyclic P2 group (22e and 22f) led to an encouraging improvement in subtype selectivity, with activity against cathepsin B being completely abolished. Reductions in activity against cathepins S and K were also observed, with a 68-fold improvement for compound 22f versus 18n against cathepsin S. Use of the cyclohexylmethyl P2 afforded a disappointing series of results when assessing selectivity. ?,? This larger P2 group needs nothing to reduce activity against cathepsin S or B, with only a modest 3–4-fold improvement in activity against cathepsin K (Table).
5: Screening Results for Compounds against Recombinant MPro Using 20 μM QS1 (Ac-Abu-Tle-Leu-Gln-ACC) as the Substrate and Selectivities Assessed against Human Recombinant Cathepsin K, B and L in the Presence of a Fluorogenic Substrate (Z-Phe-Arg-AMC for Cat K, L, and B and Z-Val-Val-Arg-AMC for Cat S)
The most active compounds from the selectivity study (22e, 22f, 23c, and 23d) were selected for additional study in the SARS-CoV-2 VeroE6 antiviral assay (Table). All four compounds were found to actively block SARS replication in a dose-dependent manner without causing any observable toxicity despite the selectivity concerns (Table). Use of the cyclohexylmethyl P2 group (23c and 23d) afforded comparable activity to 18n, with activity being observed in the micromolar range. Use of compounds with the [3.1.0] bicyclic P2 (22e and 22f), even though they were tested as mixtures of rotamers, displayed activity in the 200–300 nM range and were comparable with nirmatrelvir, which displays activity between 100 and 180 nM in the same assay. To further profile the lead compounds, solubility and metabolic screening was performed to assess their suitability for in vivo testing. All three of the tested inhibitors (18n, 22e, and 22f) were found to have acceptable k sol values, although 18n was outside of the lower limit for inhaled drug delivery (Table). All three compounds were observed to have less than desirable stability in both mouse and human microsomes. These data highlighted the need for further inhibitor optimization prior to in vivo testing, including additional exploration of proline isosteres in P2 comparable to those reported by Liu and co-workers to improve metabolic stability, while maintaining solubility values observed 22e and 22f.? In addition, further SAR exploration of the P3 biphenyl region in combination with alternative P2 groups will need to be completed to improve the overall suitability of this scaffold for in vivo testing.
6: Results for 22e, 22f, 23c, and 23d in the SARS-CoV-2 VeroE6 Antiviral Assay
Conclusions
To address both outbreaks from the most recent pandemic and any future SARS outbreaks, the development of COVID-19 antivirals against highly conserved druggable targets is crucial. M^Pro^ inhibition hinders viral replication, reducing viral load and disease severity. In this study, a potent series of covalent small-molecule biphenyl benzamide SARS-CoV-2 M^Pro^ inhibitors were synthesized and tested in both a biochemical assay against recombinant SARS-CoV-2 M^Pro^ and a virus replication assay in VeroE6-GFP cells. To identify a novel series of M^Pro^ inhibitors for this study, we initiated a scaffold hopping exercise in late 2020, based on our experience in developing small-molecule cysteine protease inhibitors against legumain. From this study, we identified a P3 biphenyl-based compound (6a) that displayed submicromolar biochemical potency in the assay developed by Drag and co-workers. This assay was reported to be 30-fold more stringent than the readily used FRET assay, which provided significant confidence in the validity of this compound. Initial SAR exploration of this scaffold highlighted the importance of the P2 leucine group to afford inhibitors with submicromolar biochemical potency. Four inhibitors (6e, 6f, 6j, and 6l) were taken forward for screening in the SARS-CoV-2 (Omicron) VeroE6-GFP antiviral assay. However, the potency observed was disappointing, with all compounds having EC_50_ values in excess of 10 μM. Further exploration of the P3 biphenyl group to improve cellular potency revealed an ortho-substitution effect. Moving from o-hydrogen to o-fluoro to o-chloro (6d, 18a, and 18n) led to stepwise improvements in both biochemical and cellular potency, with 18r being the most potent. X-ray crystallography of these inhibitors within the M^Pro^ binding site revealed that incorporation of the ortho substituent had an impact on the binding of the P1 cyclic glutamate group by strengthening the binding interactions, which was unexpected. This led to compound 18n, which displayed low-micromolar antiviral activity without toxicity. However, despite improvements in biochemical and antiviral potency via the incorporation of the o-chloro group on the P3 biphenyl, these inhibitors lacked selectivity against a panel of cysteine proteases, which was of significant concern. Further exploration of the P2 group in combination with the optimized P3 biphenyls led to the identification of compound 22e, which displayed improved antiviral activity and selectivity compared to 18n. Compound 22e, which was afforded as an inseparable mixture of rotamers around the P2–P3 amide group, exhibited a potency in the cell-based antiviral assay comparable to that of the Pfizer compound nirmatrelvir. Further exploration is required to overcome the chemistry issues associated with 22e. However this inhibitor, while demonstrating good potency in biochemical and cell-based assays with modest selectivity profiles, although it has poor metabolic stability, represents a reasonable starting point for the development of a new agent in the fight against COVID-based infections upon further optimization of the inhibitor scaffold.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Zhu Z.Lian X.Su X.Wu W.Marraro G.Zeng Y.From SARS and MERS to COVID-19: a brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses Respir. Res.202021122410.1186/s 12931-020-01479-w 32854739 PMC 7450684 · doi ↗ · pubmed ↗
- 2Li H.Liu S. M.Yu X. H.Tang S. L.Tang C. K.Coronavirus disease 2019 (COVID-19): Current status and future perspectives Int. J. Antimicrob. Agents 202055510595110.1016/j.ijantimicag.2020.10595132234466 PMC 7139247 · doi ↗ · pubmed ↗
- 3Brant A. C.Tian W.Majerciak V.Yang W.Zheng Z. M.SARS-Co V-2: from its discovery to genome structure, transcription, and replication Cell Biosci.20211113610.1186/s 13578-021-00643-z 34281608 PMC 8287290 · doi ↗ · pubmed ↗
- 4Badua C. L. D. C.Baldo K. A. T.Medina P. M. B.Genomic and proteomic mutation landscapes of SARS-Co V-2J. Med. Virol.2021931702172110.1002/jmv.2654832970329 PMC 7537117 · doi ↗ · pubmed ↗
- 5Yip A. J. W.Low Z. Y.Chow V. T. K.Lal S. K.Molnupiravir for COVID-19: The Mechanisms of Antiviral Activity Viruses 2022146134510.3390/v 1406134535746815 PMC 9228778 · doi ↗ · pubmed ↗
- 6Owen D. R.Allerton C. M. N.Anderson A. S.Aschenbrenner L.Avery M.Berritt S.Boras B.Cardin R. D.Carlo A.Coffman K. J.Dantonio A.Di L.Eng H.Ferre R.Gajiwala K. S.Gibson S. A.Greasley S. E.Hurst B. L.Kadar E. P.Kalgutkar A. S.Lee J. C.Lee J.Liu W.Mason S. W.Noell S.Novak J. J.Obach R. S.Ogilvie K.Patel N. C.Pettersson M.Rai D. K.Reese M. R.Sammons M. F.Sathish J. G.Singh R. S. P.Steppan C. M.Stewart A. E.Tuttle J. B.Updyke L.Verhoest P. R.Wei L.Yang Q.Zhu Y.(2021). An oral SARS-Co V-2 Mpro inhibitor clinical candidate for the treatment of CO · doi ↗ · pubmed ↗
- 7Vangeel L.Chiu W.De Jonghe S.Maes P.Slechten B.Raymenants J.AndréE.Leyssen P.Neyts J.Jochmans D.Remdesivir, Molnupiravir and Nirmatrelvir remain active against SARS-Co V-2 Omicron and other variants of concern Antiviral Res.202219810525210.1016/j.antiviral.2022.10525235085683 PMC 8785409 · doi ↗ · pubmed ↗
- 8Abdelnabi R.Foo C. S.Zhang X.Lemmens V.Maes P.Slechten B.Raymenants J.AndréE.Weynand B.Dallmeier K.Neyts J.The omicron (B.1.1.529) SARS-Co V-2 variant of concern does not readily infect Syrian hamsters Antiviral Res.202219810525310.1016/j.antiviral.2022.10525335066015 PMC 8776349 · doi ↗ · pubmed ↗