Biallelic FGF4 Variants Linked to Thoracic Dystrophy and Respiratory Insufficiency
Laura M. Watts, Esther Kinning, Donald R. Latner, Marla Johnston, Jessica Patrick‐Esteve, Gregory M. Cooper, Stephen R. F. Twigg, Alistair T. Pagnamenta, Jenny C. Taylor

TL;DR
A new genetic cause for thoracic dystrophy and respiratory insufficiency is identified through FGF4 gene variants in two families.
Contribution
Biallelic FGF4 variants are newly linked to thoracic dystrophy and respiratory insufficiency in humans.
Findings
Two unrelated families with thoracic dystrophy and respiratory insufficiency have biallelic FGF4 missense substitutions.
FGF4 alterations are proposed as a new cause for thoracic dystrophy without other typical diagnostic signs.
The FGF4 gene is essential for thoracic skeleton development in mice and now implicated in human disease.
Abstract
The thoracic dystrophies are inherited skeletal conditions where abnormal embryonic development of the thoracic skeleton results in a narrow chest, pulmonary hypoplasia, and respiratory insufficiency, which can be severe or lethal. The majority of thoracic dystrophies are due to biallelic alterations in genes needed for normal ciliary function. However, despite the identification of over 20 genes as causal for the thoracic dystrophy phenotype, around 20% of patients remain without a molecular diagnosis. We present two unrelated families with a clinical diagnosis of thoracic dystrophy with associated respiratory insufficiency without a molecular diagnosis on previous genetic testing. Both harbor rare biallelic and predicted deleterious missense substitutions in FGF4, a gene known to be essential for formation of the thoracic skeleton in mice. We demonstrate that the phenotype is…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
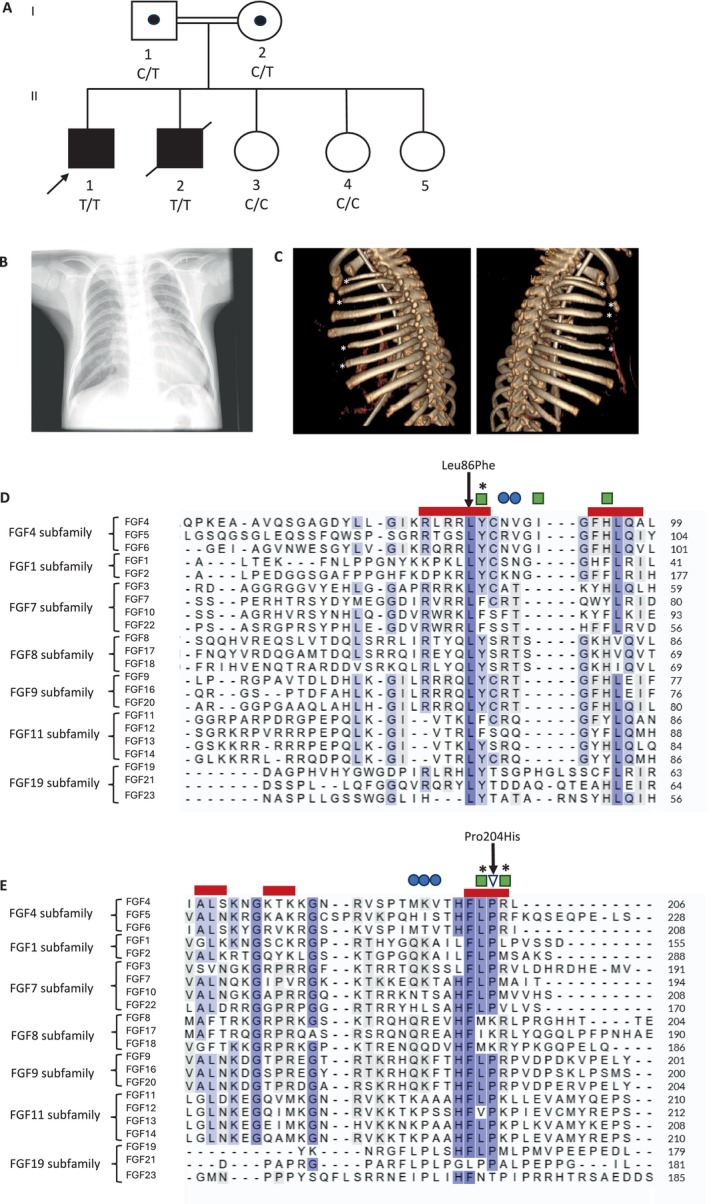 FIGURE 1
FIGURE 1| Family | Individual | Sex | Age at assessment | Antenatal findings | Neonatal resuscitation | Chest size and shape | Ribs | Spine abnormalities | Limb abnormalities | Respiratory insufficiency | Echo | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | II.1 | M | 20 years | No | Yes. | Narrow thorax | Short ribs, 11 pairs | No | No | Ventilated for first 6 months of life, then non‐invasive ventilation. Overnight ventilatory support until 20 months | Normal | Pyloric stenosis |
| II.2 | M | Deceased 3 months from respiratory infection | Yes | Small thorax | Short ribs, 11 pairs | No | No | Chronic respiratory insufficiency | Normal | NA | ||
| 2 | II.1 | M | 2 years | Small bell shaped throat, short ribs, lung volume small for gestational age | No | Thoracic dysplasia | Short ribs with hypoplasia | Secondary lumbar scoliosis | No | Pulmonary hypoplasia and chronic respiratory insufficiency, dependent upon long term mechanical ventilation via tracheostomy | PFO | Gastrostomy |
| Family | Individual | Parental consanguinity | Ethnicity | Homozygous | Exon, domain | gnomAD/UKBB frequency | CADD | AlphaMissense | SIFT | PolyPhen | REVEL |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | II.1 | Yes | Asian or British Asian: Indian |
c.256C>T p.(Leu86Phe) | 1, beta strand of FGF domain | 0/0 | 26.1 | 0.95 (likely pathogenic) | 0 (deleterious) | 1 (probably damaging) | 0.78 |
| II.2 | c.256C>T p.(Leu86Phe) | ||||||||||
| 2 | II.1 | No | European American | c.611C>A p.(Pro204His) | 3, beta strand of FGF domain | 0/0 | 29.5 | 0.99 (likely pathogenic) | 0 (deleterious) | 0.99 (probably damaging) | 0.816 |
- —National Institute for Health and Care Research10.13039/501100000272
- —National Human Genome Research Institute10.13039/100000051
- —NIHR Oxford Biomedical Research Centre10.13039/501100013373
- —Wellcome Trust10.13039/100010269
- —Medical Research Council10.13039/501100000265
- —National Cancer Institute10.13039/100000054
- —National Institute on Minority Health and Health Disparities10.13039/100006545
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCongenital Diaphragmatic Hernia Studies · Congenital Anomalies and Fetal Surgery · Neonatal Respiratory Health Research
Introduction
1
The thoracic dystrophies are a group of typically autosomal recessively‐inherited, skeletal conditions where short ribs and a narrow chest result in pulmonary hypoplasia [1, 2]. The spectrum of disease ranges from in utero or neonatal death to survival into adulthood [1]. Many of the recognized genetic causes of thoracic dystrophy involve genes essential for the function of cilia, highly specialized cellular organelles with key functions in human development [3]. While many ciliopathies have multi‐organ manifestations commonly affecting the kidney, brain, and eyes, a subset referred to as the skeletal ciliopathies are characterized by thoracic dystrophy, with or without other organ system manifestations [1]. Recognized skeletal ciliopathy syndromes include the short rib polydactyly and Jeune syndromes (DYNC2H1 and components of the ciliary intraflagellar transport pathway, among others), and Ellis‐van Creveld syndrome (EVC1 and EVC2) [1, 2].
With dedicated analysis, a genetic diagnosis can be made in the majority of patients with a thoracic dystrophy [3]. However, there remains a proportion of patients with a clinical diagnosis of thoracic dystrophy without a corresponding molecular diagnosis [1], likely representing variants in established genes that have evaded detection or novel disease‐causing genes. Novel causes of thoracic dystrophy may include genes encoding components of the ciliary apparatus not yet associated with human disease, or variants affecting other key genes and pathways involved in the formation of the rib cage.
Fibroblast growth factors (FGF) and their four corresponding tyrosine kinase fibroblast growth factor receptors (FGFR) are widely expressed in the skeleton and are essential for axial and appendicular skeletal development [4, 5, 6, 7]. Altered FGF signaling causes multiple human monogenic skeletal diseases [2, 4]. FGFs, including FGF4 and FGF8, are needed for vertebral and rib cage development [5, 6], while FGFR signaling also affects primary cilium length, including in cartilaginous growth plates and chondrocytes [8, 9]. We report two families with a clinical diagnosis of thoracic dystrophy without a molecular diagnosis. Whole genome sequencing (WGS) identified homozygous variants in FGF4, which have not been previously associated with any human disease phenotype.
Materials and Methods
2
Family 1 was recruited to the UK 100,000 Genomes Project (100KGP) for WGS under the disease category of thoracic dystrophy [10]. Family 2 was recruited for WGS through SouthSeq, a clinical research study that enrolled infants with suspected genetic disorders as part of the Clinical Sequencing Evidence‐Generating Research consortium (CSER) [11]. Variants were assessed for likely pathogenicity using in silico variant prediction tools, population allele frequencies, protein conservation, and in silico structural analysis (Supporting Information).
Results
3
Detailed case reports are available in Supporting Information.
Family 1
3.1
Family 1 (Table 1) comprises two affected boys clinically suspected to have Jeune syndrome, and three unaffected children born to healthy consanguineous 2nd cousin parents (Figure 1A). The proband (II.1) was born at term following an uncomplicated pregnancy by normal vaginal delivery. At birth, he required resuscitation for respiratory distress and was ventilated for the first 6 months of life. He had a very small thorax on clinical examination and was diagnosed with pulmonary hypoplasia. He required non‐invasive ventilation with overnight ventilatory support until 20 months old. Development was normal, and he is now 20 years old.
Pedigree, radiological images and protein sequence alignments of reported FGF4 variants. (A) Pedigree of Family 1. Colored black boxes represent individuals with a clinical diagnosis of a short rib thoracic dystrophy. Both affected children were homozygous for a missense variant in FGF4 (c.256C>T p.(Leu86Phe)). Both parents are confirmed carriers. Two of three unaffected siblings do not carry the variant. Genotypes are indicated for individuals as C (reference allele) and T (missense variant). (B) Chest x‐ray of II‐1 from Family 1 aged 3 years 6 months showing a small thorax with short ribs. Only 11 pairs of ribs are present. (C) 3D reconstruction of CT scan of II‐1 from Family 2 demonstrating hypoplasia of the 1st, 3rd, 6th and 7th ribs on the left and 1st, 3rd, 4th and 6th ribs on the right (marked with white stars). (D and E) Sequence alignment around Leu86 (D) and Pro204 (E). Amino acid sequences of all human FGFs were retrieved from Uniprot and aligned using the Clustal Omega algorithm, grouped by FGF subfamily [12]. Positions of the missense FGF4 alterations are indicated by arrows. Red boxes represent β‐strands of the FGF domain in FGF4; blue circles represent heparin binding sites; triangle represents a residue interacting with the D2–D3 linker region; residues that contact the D2 immunoglobulin domain of FGFR1 are shown by green squares; asterisks denote residues which, when altered, have reduced receptor binding and FGF activity [12, 13].
His younger brother (II.2) was born at term by vaginal delivery. At 1 h old, he was ventilated for respiratory distress. He was mildly hypotonic and had a small thorax with chronic respiratory insufficiency. He passed away at 3 months old following a respiratory infection.
X‐rays demonstrated a small thorax, short ribs, and only 11 pairs of ribs in both brothers (Figure 1B). The proband was recruited to 100KGP together with his unaffected parents and two unaffected siblings. Primary analysis using the standard 100KGP pipeline did not identify any causative variants in relevant disease panels [10].
Family 2
3.2
The proband (Table 1) is a male child aged two at last assessment, born at 35 + 1 weeks to non‐consanguineous parents. There was no relevant family history. The pregnancy was complicated by maternal methadone use, smoking, and trichomonas infection. Antenatal ultrasound showed a small thorax suspicious of a skeletal dysplasia. Fetal MRI confirmed a small bell‐shaped thorax with short ribs and low lung volume for gestational age (total 30.3 mL, < 2.5% expected, right lung 15.7 mL, left lung 14.6 mL). No prenatal genetic testing was performed.
Postnatally, his chest shape was in keeping with thoracic dystrophy and a clinical suspicion of Jeune syndrome. Chest CT demonstrated shortening and hypoplasia of the 1st, 3rd, 4th and 6th ribs on the right and 1st, 3rd, 6th and 7th ribs on the left, with otherwise normal lungs (Figure 1C). An echocardiogram demonstrated a patent foramen ovale with left‐to‐right shunting, with normal left ventricular size and systolic function. Aged two, he is dependent upon long‐term mechanical ventilation via a tracheostomy for pulmonary hypoplasia. He has impairments in speech, language, and oral motor skills, is fed via a gastrostomy, and has lumbar scoliosis. Chromosomal microarray was normal. No pathogenic variants were identified on a skeletal ciliopathies gene panel, only a heterozygous variant of uncertain significance in the recessive gene WDR60.
Genetic Analysis
3.3
All three affected individuals from the two unrelated families harbored homozygous missense variants in FGF4. In Family 1, further research analysis of musculoskeletal cases recruited to 100KGP (Supporting Information) identified a homozygous missense variant in FGF4 NM_002007.4:c.256C>T p.(Leu86Phe), absent from two unaffected siblings and present in a heterozygous state in both parents. Sanger sequencing on stored DNA from the deceased similarly affected sibling demonstrated the same homozygous FGF4 missense variant (S1A). Research WGS in Family 2 demonstrated a biallelic missense variant in FGF4 NM_002007.4:c.611C>A p.(Pro204His). Parents were both heterozygous carriers.
Both missense variants are considered to be deleterious by multiple in silico tools, with CADD scores of > 25 and annotated by AlphaMissense as likely pathogenic (Table 2) and are absent from the gnomAD database [14]. Present in gnomAD are two individuals with a different missense substitution at the same position as the Family 1 variant (p.(Leu86Pro)) in the heterozygous state where no phenotype would be expected, and 24 individuals homozygous for common polymorphisms in FGF4, which, in contrast to our variants, have little evidence for pathogenicity (Supporting Information).
Leu86 and Pro204 are both highly conserved. At position 86, there is a Leucine in all 22 human FGFs (Figure 1D) and in multiple species, including invertebrates (S1B). At position 204, there is a proline in the majority of human FGFs (Figure 1E) and vertebrates, but with greater diversity in invertebrates, where there are fewer paralogs (S1C).
Both FGF4 missense alterations are found in β strands of the core FGF domain responsible for binding to the FGF receptor [13], and are brought into close proximity in the 1.80 Å 3D protein structure (S1D). Leu86 lies immediately adjacent to Tyr87, which contacts the D2 immunoglobulin‐like domain of FGFR1 and which, when altered, results in reduced receptor binding and FGF activity [13] (Figure 1D). Leucine at position 86 is a hydrophobic amino acid buried in the core of the FGF4 protein. Alteration to the bulkier phenylalanine is likely to disrupt the conformation and interactions of FGF4 (S2), supported by in silico modeling predictions of altered hydrogen bonding involving highly conserved FGFR‐contacting residues [13] (S2).
Proline 204 directly interacts with the D2‐D3 linker region of FGFR1 [13], which contributes to ligand binding and specificity [13, 15]. Therefore, substitution to histidine would be consistent with loss of function. Specific missense substitutions in this linker region in FGFR1, FGFR2, and FGFR3 cause Pfeiffer, Apert, and Muenke syndromes, respectively [16]. Pro204 is additionally flanked by two residues that contact the D2 domain of FGFR1, and alteration of which results in reduced receptor activity [13].
Discussion
4
We describe three individuals from two families with thoracic dystrophy and pulmonary hypoplasia, each with homozygous missense variants in FGF4 in the receptor‐binding domain. The variants are absent from gnomAD and UK Biobank [14], are predicted to be damaging by multiple in silico tools, and are immediately adjacent to receptor‐contacting residues required for normal FGF signaling [13]. Consistent with a loss‐of‐function mechanism through altered binding to FGF receptors, Pro204 has been shown to directly contact FGFR1 [13], while the extremely highly conserved Leu86 residue is mutated to a bulkier amino acid likely to compromise structural conformation.
The vertebrae and rib cage form from somites, which periodically segment from the presomitic mesoderm in a process requiring FGF4 [5, 17]. Mice lacking Fgf4 alone or in combination with Fgf8 demonstrate a spectrum of vertebral and rib defects resulting from abnormal somitogenesis with similarities to the phenotypes described in our patients, including missing ribs and an abnormally shaped thoracic cage [6, 17]. In mice, inactivation of Fgf4 in the presomitic mesoderm has also been shown to result in abnormal patterns of expression of both Mesp2 and Hes7 [17]. Human pathogenic alterations in MESP2 and HES7 cause spondylocostal dysostosis characterized by rib and vertebral abnormalities [18]. Nevertheless, despite the important role of FGF4 in somitogenesis, formal confirmation of the pathogenicity of the described variants requires dedicated functional studies such as study of variant‐specific model organisms, in vitro assessment of mutant FGF4‐receptor binding, and investigation of ciliary signaling pathways. Identification of additional affected families would further support this work.
Many ciliopathies have multiple organ manifestations resulting from diverse ciliary functions during development [1, 3]. In contrast, the families reported had a phenotype largely restricted to thoracic dystrophy. Although FGF receptor signaling has been shown to be involved in the regulation of cilium length [8, 9], and in zebrafish Fgf4 may be needed for normal cilia development [19], FGF4 is not well established as an essential ciliary gene in humans or mice. The phenotype reported does not include limb shortening or characteristic radiological features such as trident acetabulum found in other thoracic dystrophies [1], nor characteristic clinical or radiological features of Kagami‐Ogata syndrome. In mice, Fgf4 is known to be expressed in the apical ectodermal ridge responsible for normal limb bud outgrowth [7, 20], functional redundancy with Fgf8 means that loss of Fgf4 alone does not result in a limb phenotype [7, 20].
Clinically, we have been able to provide a likely diagnosis for the deceased sibling in Family 1, and reproductive counseling for his surviving 20‐year‐old brother. Identification of novel rare disease genes allows expansion of genetic testing panels and biological insights which may aid research into specific treatments. Our work suggests that homozygous variants in FGF4 may lead to a phenotype of thoracic abnormalities with respiratory insufficiency, which could prompt screening of other rare disease cohorts, or inclusion in diagnostic testing panels.
Author Contributions
Laura M. Watts, Alistair T. Pagnamenta, Jenny C. Taylor: study conception. All authors: data acquisition. Laura M. Watts: manuscript initial drafting. All authors: manuscript revision, editing, and approval.
Ethics Statement
Ethics approval for the 100,000 Genomes Project was from Cambridge South REC (14/EE/1112). The review board at the University of Alabama at Birmingham (IRB‐300000328) approved and monitored the SouthSeq study (family 2).
Consent
All patients or their legal guardians gave signed informed consent for publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Data S1. Supporting Information.
Data S2. Supporting Information.
Data S3. Supporting Information.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1W. Zhang , S. P. Taylor , H. A. Ennis , et al., “Expanding the Genetic Architecture and Phenotypic Spectrum in the Skeletal Ciliopathies,” Human Mutation 39 (2018): 152–166.29068549 10.1002/humu.23362 PMC 6198324 · doi ↗ · pubmed ↗
- 2S. Unger , C. R. Ferreira , G. R. Mortier , et al., “Nosology of Genetic Skeletal Disorders: 2023 Revision,” American Journal of Medical Genetics. Part A 191 (2023): 1164–1209.36779427 10.1002/ajmg.a.63132 PMC 10081954 · doi ↗ · pubmed ↗
- 3A. Hammarsjö , M. Pettersson , D. Chitayat , et al., “High Diagnostic Yield in Skeletal Ciliopathies Using Massively Parallel Genome Sequencing, Structural Variant Screening and RNA Analyses,” Journal of Human Genetics 66 (2021): 995–1008.33875766 10.1038/s 10038-021-00925-x PMC 8472897 · doi ↗ · pubmed ↗
- 4D. M. Ornitz and P. J. Marie , “Fibroblast Growth Factor Signaling in Skeletal Development and Disease,” Genes and Development 29 (2015): 1463–1486.26220993 10.1101/gad.266551.115PMC 4526732 · doi ↗ · pubmed ↗
- 5L. A. Naiche , N. Holder , and M. Lewandoski , “FGF 4 and FGF 8 Comprise the Wavefront Activity That Controls Somitogenesis,” Proceedings of the National Academy of Sciences of the United States of America 108 (2011): 4018–4023.21368122 10.1073/pnas.1007417108 PMC 3054031 · doi ↗ · pubmed ↗
- 6A. M. Boulet and M. R. Capecchi , “Signaling by FGF 4 and FGF 8 Is Required for Axial Elongation of the Mouse Embryo,” Developmental Biology 371 (2012): 235–245.22954964 10.1016/j.ydbio.2012.08.017PMC 3481862 · doi ↗ · pubmed ↗
- 7F. V. Mariani , C. P. Ahn , and G. R. Martin , “Genetic Evidence That FG Fs Have an Instructive Role in Limb Proximal‐Distal Patterning,” Nature 453 (2008): 401–405.18449196 10.1038/nature 06876 PMC 2631409 · doi ↗ · pubmed ↗
- 8J. M. Neugebauer , J. D. Amack , A. G. Peterson , B. W. Bisgrove , and H. J. Yost , “FGF Signalling During Embryo Development Regulates Cilia Length in Diverse Epithelia,” Nature 458 (2009): 651–654.19242413 10.1038/nature 07753 PMC 2688717 · doi ↗ · pubmed ↗