16S rRNA amplicon sequencing of bacterial communities in Solid Waste Leachates (SWL) from Olusosun Dumpsite, Ojota, Lagos State, Nigeria
Adewale K. Ogunyemi, Olanike M. Buraimoh, Bukola C. Ogunyemi, Titilola A. Samuel, Matthew O. Ilori, Olukayode O. Amund

TL;DR
This study analyzes bacterial communities in leachates from a Nigerian dumpsite using 16S rRNA sequencing to inform waste management strategies.
Contribution
The study provides a detailed bacterial community profile of leachates from a specific Nigerian dumpsite using 16S rRNA sequencing.
Findings
Acidobacteria was the most abundant phylum in the leachates, at 14.65%.
The findings offer insights into microbial composition that could guide waste management practices.
Abstract
Here, we use 16S rRNA gene sequencing to identify bacterial community structure of solid waste leachates from Olusosun dumpsite. Acidobacteria (14.65 %) was the most abundant phylum with clear affiliations. This was followed by Planctomycetes (7. 15 %), Proteobacteria (3.28 %), Chloroflexi (1.41 %), Actinobacteria (0.91 %) and other phyla (0.96 %). Data obtained provides valuable insight into potential strategies for waste management.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
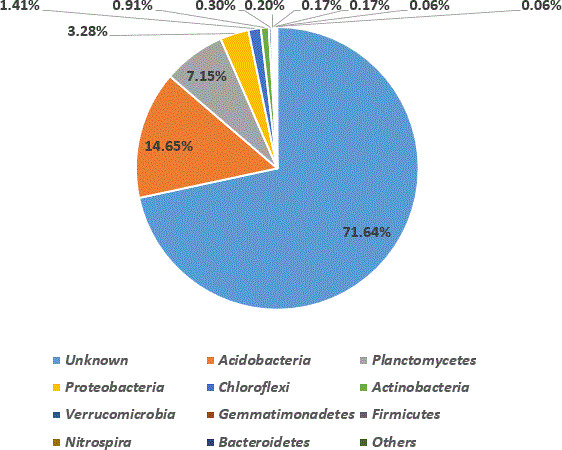 Fig 1
Fig 1| Parameter | Value |
|---|---|
| Total reads (bp) | 296,086 |
| Average read length (bp) | 173,888 |
| G + C (%) | 57.86 |
| Coverage | 68.0031 |
| Size of homopolymer | 3 |
| OTU length (bp) | 469 |
| OTU total count | 145 |
| Inverse Simpson index | 0.989 |
| Shannon’s evenness average | 6.638 |
| Chao1 richness estimator | 256.272 |
| Abundance-based coverage estimator | 258.781 |
| Good coverage | 0.441 |
| Simpson | 0.989 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial Community Ecology and Physiology · Genomics and Phylogenetic Studies · Gut microbiota and health
ANNOUNCEMENT
Recent times have seen a rise in the threat of dumpsite contamination due to leachate produced from solid waste disposal, which is strongly influenced by the waste composition, volume of leachate generated, and distance from water bodies (1). The present study aimed to assess the bacterial community structure and diversity in solid waste leachates at Olusosun dumpsite, Ojota, Lagos, Nigeria. The decomposing solid waste was collected from Olusosun dumpsite (coordinates: N 6°29′21.8″; E 003°23′29.3″). Olusosun dumpsite is the most active of all dumpsites in Lagos State (2), the most populous city in sub-Saharan Africa.
The genomic DNA was obtained using the ZR Fungal/Bacterial DNA Kit (Zymo Research, Irvine, CA, USA) following the manufacturer’s instructions. The genomic DNA samples were amplified using universal primers 341F (5′-CCTACGGGNGGCWGCAG) and 785R (5′-. GACTACHVGGGTATCTAATCC) that target the V3–V4 region of the 16S rRNA gene (3). PCR program was run as follows: initial denaturation at 95°C for 3 min, followed by 30 cycles of denaturation at 95°C for 30 s, annealing at 56°C for 30 s, elongation at 72°C for 1 min, and a final elongation step at 72°C for 5 min.
With the use of a MiSeq v3 (600 cycles) Kit, the amplicon was sequenced on Illumina’s MiSeq platform, and about 20 Mb of data (2 × 300 bp long paired-end reads) was generated for each sample. However, Inqaba’s in-house developed data analysis pipeline was employed for BLAST-based data analysis. A standalone Ribopicker v0.4.3 (4) was utilized to remove possible non-rRNA sequences from the raw sequencing reads using the Greengene database (5). FasQC v0.11.9 (6) was used for the quality assessment of the raw amplicon reads. Pre-processing included the use of Trimmomatics v0.39 (7) for the removal of adaptor sequences, low quality (using phred33), and short reads (<100 bp) for both the paired-end files.
Post-assessed reads were further processed using QIIME2 v2020.6.0 (8) for sequence denoising methods. The denoising method was implemented through the use of DADA2 (QIIME2 plugin:q2-DADA2) (9) with truncating lengths of 284 and 118 for the forward and reversed reads, respectively. Instead of grouping high-quality sequences at a 97 % similarity threshold into operational taxonomic units (OTU), we based on amplicon sequence variants (ASVs), which utilize DNA directly for taxonomic placements instead of clustering of closely related individuals at a 97 % similarity threshold. ASVs were implemented into qiime2 and trained using three databases for comparative taxonomic profiling: SILVA (10), Greengene (5), and Ribosomal Database Project (11). The overall raw sequencing data had 296,086 total reads and an average read length of 173,888 base pairs with 57.86% guanine + cytosine content (Table 1). Fig. 1 depicts taxa relative abundance of major bacterial phyla (>0.05 %). The bacterial communities had an unclear phylum affiliation of 71.64 % abundance. Other bacterial phyla with an abundance ≥ 0.05 % included Acidobacteria (14.65 %), Planctomycetes (7.15 %), Proteobacteria (3.28 %), Chloroflexi (1.41 %), Actinobacteria (0.91 %), Verrucomicrobia (0.30 %), Gemmatimonadetes (0.20 %), Firmicutes (0.17 %), Nitrospira (0.17 %), Bacteroidetes (0.06 %), and others (0.06 %) (Fig. 1).
Taxa relative abundance and taxa taxonomic composition of OTUs at the phylum level.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Slomczynska B, Slomczynski T. 2004. Physicochemical and toxicological characteristics of leachates from MSW landfills. Polish Journal of Environmental Studies 13:627–637.
- 2Okafor CL, Chukwu FS, Ahove MA. 2024. Comparative assessment of air quality in Epe, and Olusosun dumpsites of Nigeria. J Agri Environ Sci 9:50–60. doi:10.4314/jaes.v 9i 1.4 · doi ↗
- 3Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO. 2013. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:e 1. doi:10.1093/nar/gks 80822933715 PMC 3592464 · doi ↗ · pubmed ↗
- 4Schmieder MR, Lim YW, Edwards R. 2012. Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics 28:433–435. doi:10.1093/bioinformatics/btr 66922155869 PMC 3268242 · doi ↗ · pubmed ↗
- 5De Santis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S r RNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. doi:10.1128/AEM.03006-0516820507 PMC 1489311 · doi ↗ · pubmed ↗
- 6Andrews S. 2010. Fast QC: a quality control tool for high throughput sequence data,. Babraham Bioinformatics. Babraham Institute, Cambridge, United Kingdom.
- 7Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi:10.1093/bioinformatics/btu 17024695404 PMC 4103590 · doi ↗ · pubmed ↗
- 8Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al.. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. doi:10.1038/s 41587-019-0209-931341288 PMC 7015180 · doi ↗ · pubmed ↗