Complete coding sequence of goose-derived cluster 3.2 Tembusu virus from Anhui province of China in October 2024
Lei Zhao, Jie Zhu, Shao-Jun He, Peng-Fei Ye, Jie Kong, Liu-Jun Zhang

TL;DR
A new strain of Tembusu virus was identified in a goose from China in 2024 and classified into a specific genetic group.
Contribution
The complete coding sequence of a novel goose-derived TMUV strain from China is reported and classified into Cluster 3.2.
Findings
The AHWH isolate shares the highest nucleotide identity with TMUV/Goose/CHN/2022/HQ-22.
The virus was classified into TMUV Cluster 3.2 based on phylogenetic analysis.
The isolate was derived from a goose with pancreatic necrosis in Anhui province.
Abstract
In October 2024, a novel circulating Tembusu virus (TMUV) strain was isolated and characterized from an out-of-season breeding goose with pancreatic necrosis from Anhui province of China. Phylogenetic analysis revealed that the AHWH isolate shared the highest nucleotide identity with goose-derived TMUV/Goose/CHN/2022/HQ-22 and was classified into TMUV Cluster 3.2.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
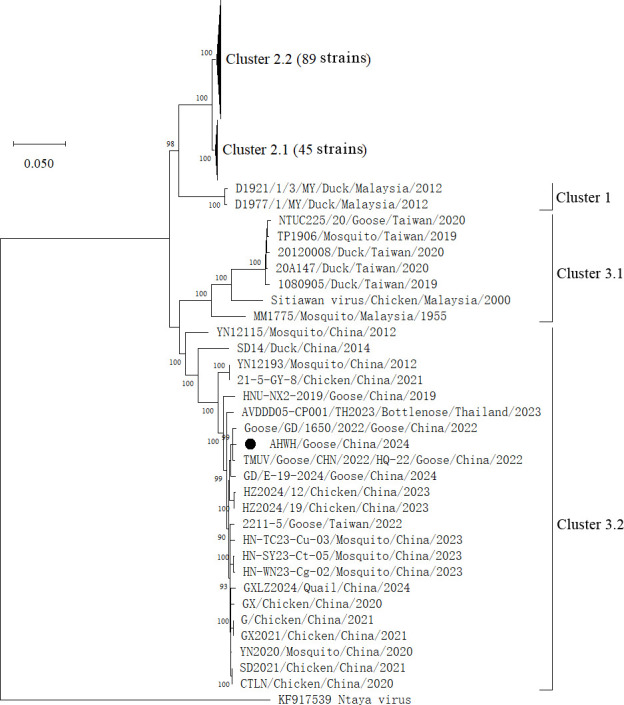 Fig 1
Fig 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMosquito-borne diseases and control · Viral Infections and Vectors · Virology and Viral Diseases
ANNOUNCEMENT
Tembusu virus (TMUV) belongs to the genus Orthoflavivirus in the family Flaviviridae. Since 2010, TMUV has caused a severe disease characterized by a drop in egg production and neurological signs in poultry in China (1–4). Here, we report the complete coding sequence of TMUV strain AHWH isolated from an out-of-season breeding goose located in Wuhe County (33.1465°N, 117.9923°E), Anhui Province, China in October 2024.
The animal study was reviewed and approved by Anhui Science and Technology University Committee of Animal Experiments approval ID: 2025076. The AHWH strain, isolated from a 44-day-old goose that died of pancreatic necrosis and tested TMUV-positive by reverse transcription-polymerase chain reaction (RT-PCR), was recovered by standard allantoic cavity inoculation of 10-day-old specific-pathogen-free (SPF) chicken embryos (5). Viral RNA was extracted from the allantoic fluid using the MiniBEST Universal RNA Extraction Kit (TaKaRa, Beijing, China). Library preparation and Illumina sequencing were performed at Shanghai Tanpu Biotechnology Co., Ltd. (Shanghai, China). Briefly, the NEBNext Ultra II RNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA) was used to construct paired-end libraries. After adapter ligation, 10 cycles of PCR amplification were performed to enrich the sequencing target. Sequencing was performed on an Illumina NovaSeq 6000 System (Illumina, San Diego, CA, USA) to generate 150 bp paired-end reads. Raw reads were processed with Fastp (6) to remove adapters and low-quality bases (Phred score <20). Host-derived reads (including ribosomal RNAs) were removed using BBMAP v38.51 (7). De novo genome assembly was performed using SPAdes v3.14.1 (8). These extracted assembled scaffolds limited the minimum contig length to 100 bases, with the best BLAST hits to the National Center of Biotechnology Information (NCBI) nucleotide database. A total of 6,928,796 raw reads were obtained. The final consensus sequence was generated from 552,647 filtered reads with an average depth of 4.86 × 10^3^. Taxonomic identification confirmed it as TMUV through nucleotide BLAST webserver (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Subsequently, the Open Reading Frame (ORF) of this strain was aligned by the MUSCLE program in MEGA version 12 together with other selected TMUV strains from GenBank (9). A neighbor-joining phylogeny was reconstructed using the Maximum Composite Likelihood substitution model in MEGA12 with the bootstrap test (1,000 replicates). Default parameters were used for all software unless otherwise specified.
The final consensus sequence of the AHWH isolate was 11,030 bp in length with 49.18% GC content and contained a unique ORF of 10,278 bp encoding a polyprotein of 3,425 amino acids, along with a 79 bp 5′ untranslated region (UTR) and a 673 bp 3′ UTR. Compared to the TMUV/Goose/CHN/2022/HQ-22 strain (OR909676), our sequence lacked a 15 bp sequence in the 5′ UTR but had an additional 50 bp sequence in the 3′ UTR. The ORF sequence alignments revealed that the AHWH strain shared 85.27%–99.35% nucleotide identities with the published TMUV strains. Specifically, it showed the highest similarity to the goose-derived strain TMUV/Goose/CHN/2022/HQ-22 (99.35%). Phylogenetic analyses of the ORF indicated that the AHWH isolate was grouped into Cluster 3.2 (Fig. 1). Of note, the out-of-season goose farming period in Southeast China overlapped with the local mosquito activity season, thereby likely facilitating interspecies transmission of Cluster 3.2 TMUV between geese and mosquitoes (10–13).
Neighbor-joining phylogeny of TMUV based on the ORF nucleotide sequences. Reference sequences were retrieved from GenBank. Multiple sequence alignment was performed using the MUSCLE program, and the Neighbor-joining phylogenetic tree was reconstructed using the Maximum Composite Likelihood substitution model with the bootstrap test (1,000 replicates) in MEGA version 12. Only bootstrap values exceeding 80% are displayed at branch nodes. Virus strains are annotated with strain name, host, country, and year of isolation. The TMUV AHWH isolate is denoted by a solid circle. The Ntaya isolate was used as the outgroup.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Su J, Li S, Hu X, Yu X, Wang Y, Liu P, Lu X, Zhang G, Hu X, Liu D, Li X, Su W, Lu H, Mok NS, Wang P, Wang M, Tian K, Gao GF. 2011. Duck egg-drop syndrome caused by BYD virus, a new Tembusu-related flavivirus. P Lo S One 6:e 18106. doi:10.1371/journal.pone.001810621455312 PMC 3063797 · doi ↗ · pubmed ↗
- 2Zhu Y, Hu Z, Lv X, Huang R, Gu X, Zhang C, Zhang M, Wei J, Wu Q, Li J, Zhang R, Cao S, Yin D, Wang B, Liu G, Wang G. 2022. A novel Tembusu virus isolated from goslings in China form a new subgenotype 2.1.1. Transbound Emerg Dis 69:1782–1793. doi:10.1111/tbed.1415533993639 · doi ↗ · pubmed ↗
- 3Yu Z, Ren H, Sun M, Xie W, Sun S, Liang N, Wang H, Ying X, Sun Y, Wang Y, Zheng Y, Hu X, Su J. 2022. Tembusu virus infection in laying chickens: Evidence for a distinct genetic cluster with significant antigenic variation. Transbound Emerg Dis 69:e 1130–e 1141. doi:10.1111/tbed.1440234821052 · doi ↗ · pubmed ↗
- 4Yan D, Li X, Wang Z, Liu X, Dong X, Fu R, Su X, Xu B, Teng Q, Yuan C, Zhang Z, Liu Q, Li Z. 2022. The emergence of a disease caused by a mosquito origin cluster 3.2 Tembusu virus in chickens in China. Vet Microbiol 272:109500. doi:10.1016/j.vetmic.2022.10950035792374 · doi ↗ · pubmed ↗
- 5WOAH. 2023. Chapter 3.3.4: Avian influenza (infection with avian influenza viruses). In Manual of diagnostic tests and vaccines for terrestrial animals
- 6Chen S. 2023. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. Imeta 2:e 107. doi:10.1002/imt 2.10738868435 PMC 10989850 · doi ↗ · pubmed ↗
- 7Bushnell B. 2018. Bbtools: a suite of fast, multithreaded bioinformatics tools designed for analysis of DNA and RNA sequence data. https://jgi.doe.gov/data-and-tools/bbtools.
- 8Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A. 2020. Using SP Ades de novo assembler. Curr Protoc Bioinformatics 70:e 102. doi:10.1002/cpbi.10232559359 · doi ↗ · pubmed ↗