Draft genomes of norovirus from stool samples of under-five children presenting with gastroenteritis in Malawi from 2012 to 2024
Ernest Matambo, Flywell Kawonga, Chimwemwe Mhango, End Chinyama, Josephine Msowoya, Clara Majengo, Sesiyanda Maseko, Nkosazana Shange, Surprise Baloyi, Milton T. Mogotsi, Francis E. Dennis, Celeste Donato, Benjamin Kumwenda, Martin M. Nyaga, Chrispin Chaguza, Khuzwayo C. Jere

TL;DR
This study presents five draft norovirus genomes from young children in Malawi who had gastroenteritis between 2012 and 2024.
Contribution
The study provides new genomic data on norovirus strains circulating in Malawi over a 12-year period.
Findings
Five draft norovirus genomes were successfully sequenced from stool samples.
The samples were collected from under-five children with gastroenteritis in Malawi.
The data spans a 12-year period from 2012 to 2024.
Abstract
Norovirus is one of the most important etiological agents of gastroenteritis (GE) and food-borne diarrhea in all age groups worldwide. Here we report five draft genomes of norovirus isolated from stool samples collected from under-five children presenting with GE in Malawi from 2012 to 2024.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
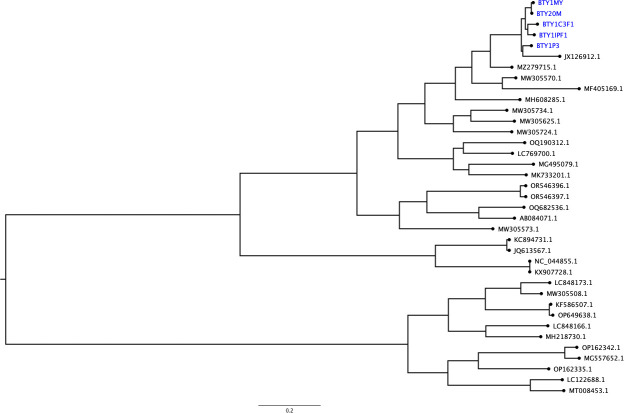 Fig 1
Fig 1| BTY1P3 | BTY1C3F1 | BTY1IPF1 | BTY1MY | BTY20M | |
|---|---|---|---|---|---|
| Sample | Stool | Stool | Stool | Stool | Stool |
| Date of collection | 26 September 2016 | 19 June 2015 | 05 October 2016 | 11 August 2016 | 23 August 2017 |
| Ct values | 26.379 | 28.448 | 23.855 | 28.454 | 34.332 |
| Sequencing platform | Illumina NextSeq 2000 | Illumina NextSeq 2000 | Illumina NextSeq 2000 | Illumina NextSeq 2000 | Illumina NextSeq 2000 |
| Reads | Paired | Paired | Paired | Paired | Paired |
| Average read length (trimmed) | 119.21 | 120.41 | 128.47 | 124.53 | 124.17 |
| Average sequencing depth | 267.3× | 754.3× | 992.9× | 183.3× | 1115.8× |
| Average sequencing coverage (%) | 99.9 | 97.8 | 99.9 | 98.3 | 99.7 |
| Reference |
|
|
|
|
|
| GC (%) | 49.84 | 49.47 | 49.99 | 49.41 | 49.35 |
| Assembly length | 7,496 | 7,379 | 7,547 | 7,420 | 7,524 |
| Completeness (%) | 99.1 | 97.55 | 99.77 | 98.1 | 99.47 |
| Sequence identity (%) | 95.7 | 91.4 | 94.7 | 92.5 | 94.3 |
| Genogroup | GII | GII | GII | GII | GII |
- —Bill and Melinda Gates Foundationhttp://dx.doi.org/10.13039/100000865
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsViral gastroenteritis research and epidemiology · Bacteriophages and microbial interactions · Virus-based gene therapy research
ANNOUNCEMENT
Despite being a single-stranded RNA virus, norovirus is stable, highly infectious, and has been associated with gastroenteritis (GE) and food-borne diarrhea in all age groups globally (1–3). Norovirus (genus, Norovirus and family, Caliciviridae) has an approximately 7.5 kb genome with three open reading frames that encode a polyprotein (~5,100 bp), a viral protein 1 (~1,600 bp), and a viral protein 2 (~720 bp) (4–6). There are 10 known norovirus genogroups: GI–GX (7). Genogroups GI and GII primarily infect humans, whereas GIV, GVIII, and GIX rarely infect humans (3, 4, 7). In Malawi, norovirus is the third most prevalent viral etiological agent of GE but data on its genomic characterization are unavailable (8, 9). As part of the Sequencing and Antigenic Cartography of Enteric Viruses project, we randomly selected 10 archived stool samples per month from 2012 to 2024 that were collected from children of age less than 5 years who presented with GE at Queen Elizabeth Central Hospital for norovirus detection and sequencing. We report five draft genome sequences of the norovirus-positive RNA extracts that were successfully sequenced.
RNA was extracted from stool samples using the QIAamp Fast DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Norovirus was detected through real-time polymerase chain reaction using custom-designed enteric TaqMan Array Cards in methods as previously described (8, 10). The norovirus-positive stool samples were re-extracted using QIAamp RNA Mini kit (Qiagen, Hilden, Germany) and then quantified on a Qubit fluorometer using a High Sensitivity ssRNA Assay kit (Thermo Fisher Scientific, USA). cDNA was synthesized through whole transcriptome amplification using a Qiagen FX Whole Transcriptome Amplification kit. Genomic libraries were prepared using the Illumina DNA Prep kit (Illumina, USA) before being sequenced on Illumina NextSeq 2000 platform using a P1 flow cell and 300-cycle reagent kit (2 × 150 bp paired-end reads).
Reads were quality assessed using FastQC 0.11.7 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), trimmed using Trimmomatic 0.39, and their quality parameters collated using MultiQC 1.27.1 (11, 12). Contaminating human reads were removed by mapping to the human reference genome (GRCh38.p13) using Bowtie v2.5.4 (13). The non-human reads were mapped to the reference genome (JX459908.1) using the Burrows-Wheeler Alignment 0.7.18-r1243-dirty and variant calling was done using iVar 1.4.4 (14, 15). The assemblies were identified as norovirus using Genome Detective Virus Tool, genogroups assigned using a genotyping tool (https://www.rivm.nl/mpf/typingtool/norovirus/) and annotated using Prokka 1.14.6 (16, 17). Assembly completeness and identity were calculated using CheckV 1.0.3 and needle in EMBOSS 6.6.0.0 (http://emboss.open-bio.org/) respectively (18). Table 1 and Fig. 1 summarize the assembly characteristics and illustrate the phylogeny of the assemblies and the global genomes. The assemblies showed low divergence among the strains, possibly due to recent evolution from a common ancestor.
Maximum likelihood phylogenetic tree of 1% of 3,683 human norovirus global sequences and the five Malawi draft norovirus genomes (in blue, BTY1C3F1: PV611445, BTY1IPF1: PV611446, BTY1MY: PV611447, BTY1P3: PV611448, BTY20M: PV611449) subsampled based on phylogenetic diversity using Environment for Tree Exploration 3.1.3 (ETE3) toolkit (19). The global sequences were downloaded from NCBI using data sets 18.1.0 (https://www.ncbi.nlm.nih.gov/datasets/docs/v2/command-line-tools/). Multiple sequence alignment was performed using MAFFT ver.7, alignment curation was performed using trimAl v1.5.rev0 using automated parameters and visualized in Seaview 5.0.5 (20–22). The phylogenetic tree was constructed using IQ-TREE 2.3.6 with 1,000 bootstraps, visualized in FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) and was midpoint rooted (23). The Malawi genomes clustered with published GII.17 and GII.4 strains (MZ279715.1 and JX126912.1, respectively) from the USA, supporting their GII genogroup classification.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hjort RG, Pola CC, Soares RRA, Oliveira DA, Stromberg L, Claussen JC. 2024. Advances in biosensors for detection of foodborne microorganisms, toxins, and chemical contaminants, p 372–384. In Encyclopedia of food safety. Elsevier.
- 2Parra GI. 2019. Emergence of norovirus strains: a tale of two genes. Virus Evol 5:vez 048. doi:10.1093/ve/vez 04832161666 PMC 6875644 · doi ↗ · pubmed ↗
- 3Chen D, Li Y, Lv J, Liu X, Gao P, Zhen G, Zhang W, Wu D, Jing H, Li Y, Zhao Y, Ma X, Ma H, Zhang L. 2019. A foodborne outbreak of gastroenteritis caused by Norovirus and Bacillus cereus at a university in the Shunyi District of Beijing, China 2018: a retrospective cohort study. BMC Infect Dis 19:910. doi:10.1186/s 12879-019-4570-631664944 PMC 6819576 · doi ↗ · pubmed ↗
- 4Chan M, Kwan HS, Chan P. 2017. Structure and genotypes of noroviruses. Norovirus. doi:10.1016/B 978-0-12-804177-2.00004-X · doi ↗
- 5Lo M, Doan YH, Mitra S, Saha R, Miyoshi S-I, Kitahara K, Dutta S, Oka T, Chawla-Sarkar M. 2024. Comprehensive full genome analysis of norovirus strains from Eastern India, 2017-2021. Gut Pathog 16:3. doi:10.1186/s 13099-023-00594-538238807 PMC 10797879 · doi ↗ · pubmed ↗
- 6Tohma K, Lepore CJ, Martinez M, Degiuseppe JI, Khamrin P, Saito M, Mayta H, Nwaba AUA, Ford-Siltz LA, Green KY, Galeano ME, Zimic M, Stupka JA, Gilman RH, Maneekarn N, Ushijima H, Parra GI. 2021. Genome-wide analyses of human noroviruses provide insights on evolutionary dynamics and evidence of coexisting viral populations evolving under recombination constraints. P Lo S Pathog 17:e 1009744. doi:10.1371/journal.ppat.100974434255807 PMC 8318288 · doi ↗ · pubmed ↗
- 7Doh H, Lee C, Kim NY, Park Y-Y, Kim E-J, Choi C, Eyun S-I. 2025. Genomic diversity and comparative phylogenomic analysis of genus norovirus. Sci Rep 15:5412. doi:10.1038/s 41598-025-87719-939948168 PMC 11825734 · doi ↗ · pubmed ↗
- 8Iturriza-Gómara M, Jere KC, Hungerford D, Bar-Zeev N, Shioda K, Kanjerwa O, Houpt ER, Operario DJ, Wachepa R, Pollock L, Bennett A, Pitzer VE, Cunliffe NA. 2019. Etiology of diarrhea among hospitalized children in Blantyre, Malawi, following rotavirus vaccine introduction: a case-control study. J Infect Dis 220:213–218. doi:10.1093/infdis/jiz 08430816414 PMC 6581894 · doi ↗ · pubmed ↗