ERα36 Promotes MDR1-Mediated Adriamycin Resistance via Non-Genomic Signaling in Triple-Negative Breast Cancer
Muslimbek Mukhammad Ugli Poyonov, Anh Thi Ngoc Bui, Seung-Yeon Lee, Gi-Ho Lee, Hye-Gwang Jeong

TL;DR
This study shows that ERα36 helps breast cancer cells resist chemotherapy by boosting MDR1 through non-genomic signaling, offering a new target for treatment.
Contribution
The paper reveals a novel non-genomic mechanism by which ERα36 promotes Adriamycin resistance via MDR1 upregulation in triple-negative breast cancer.
Findings
ERα36 activation increases MDR1 expression and drug resistance in breast cancer cells.
ERα36 activates Akt/ERK-NF-κB/CREB and Wnt/β-catenin pathways to upregulate MDR1.
Silencing ERα36 reduces MDR1 expression and drug resistance.
Abstract
Drug resistance remains a critical barrier to effective treatment in several cancers, particularly triple-negative breast cancer (TNBC). Estrogen receptor α36 (ERα36), a variant of the estrogen receptor in ER-negative breast cancer cells, plays important roles in cancer cell proliferation. We investigated the role of ERα36 in regulating multidrug resistance protein 1 (MDR1) in MDA-MB-231 human breast cancer cells. The activation of ERα36 by BSA-conjugated estradiol (BSA-E2) increased cell viability under Adriamycin exposure, suggesting its involvement in promoting drug resistance. BSA-E2 treatment significantly reduced the intracellular rhodamine-123 levels by activating the MDR1 efflux function, which was linked to increased MDR1 transcription and protein expression. The mechanical ERα36-mediated BSA-E2-induced activation of EGFR and downstream signaling via c-Src led to an activation…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
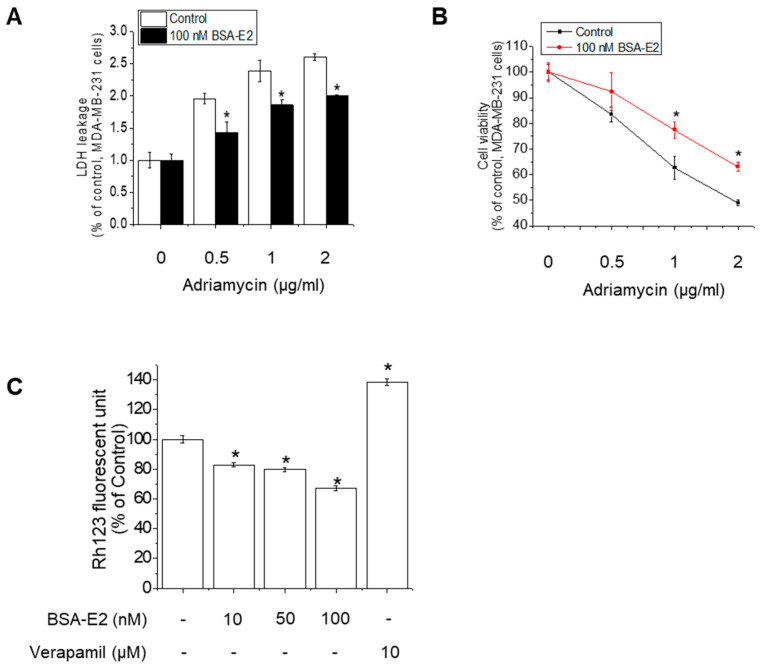 Figure 1
Figure 1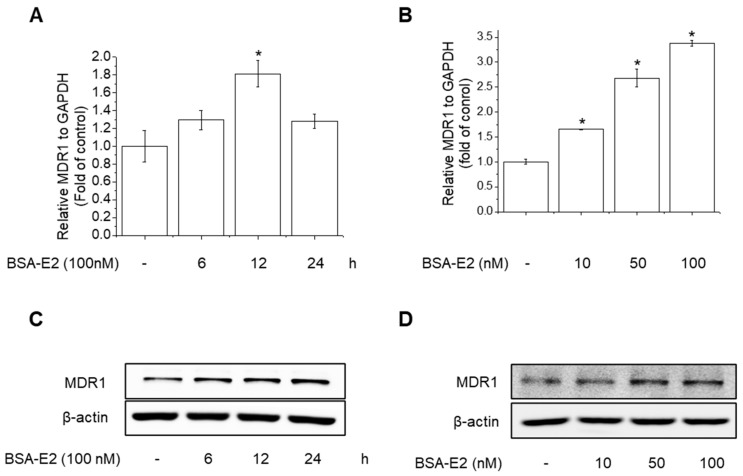 Figure 2
Figure 2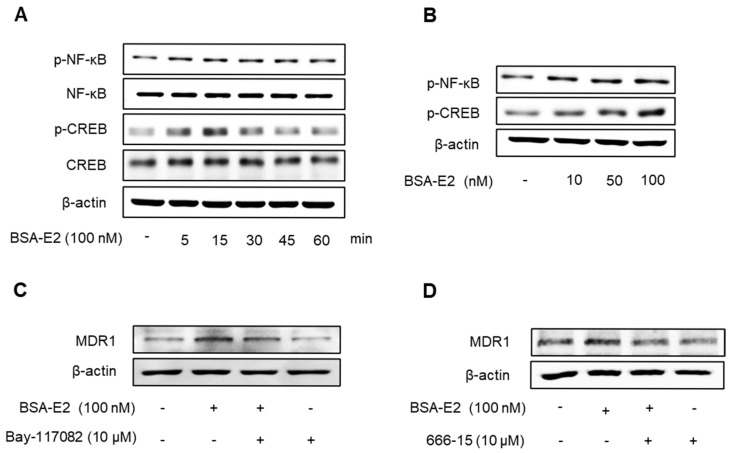 Figure 3
Figure 3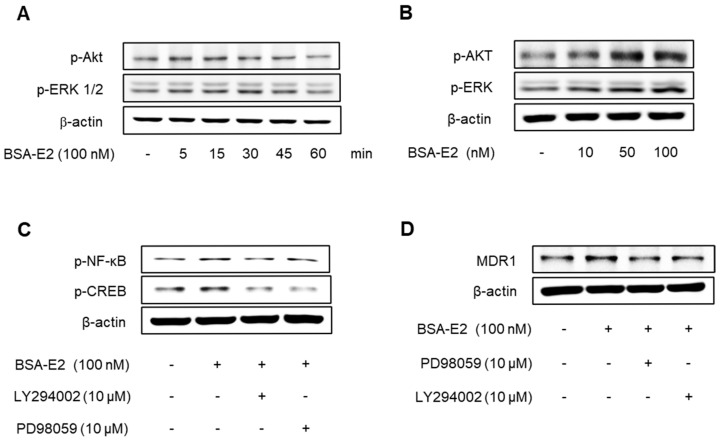 Figure 4
Figure 4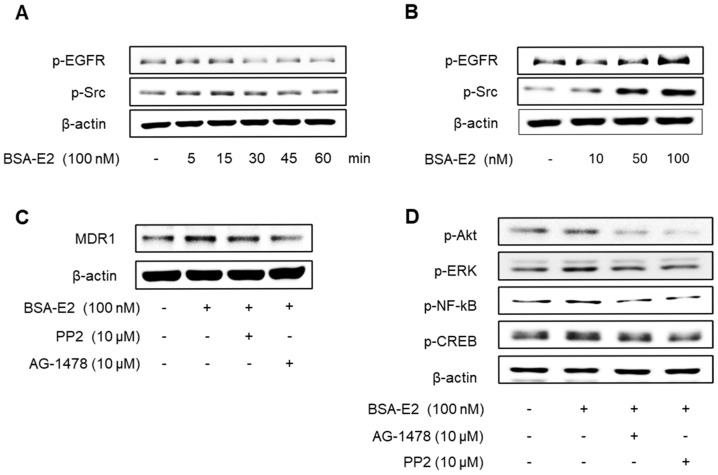 Figure 5
Figure 5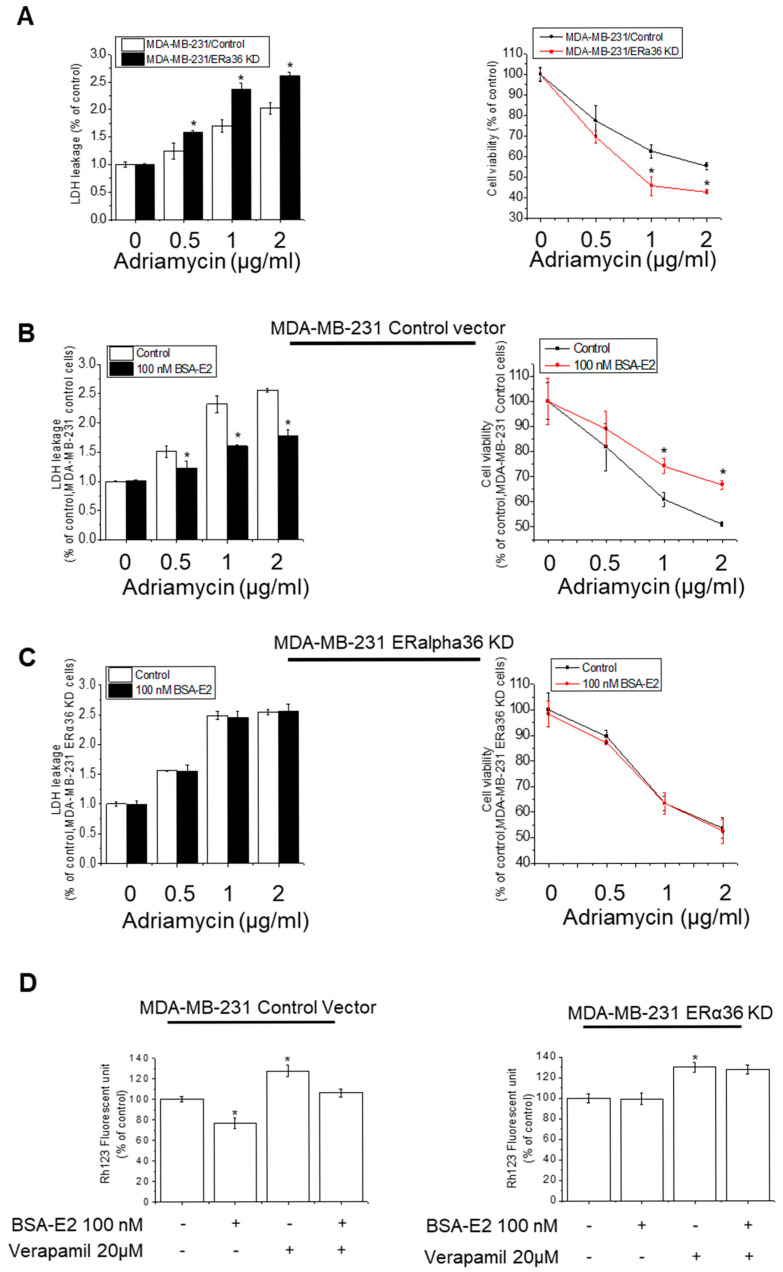 Figure 6
Figure 6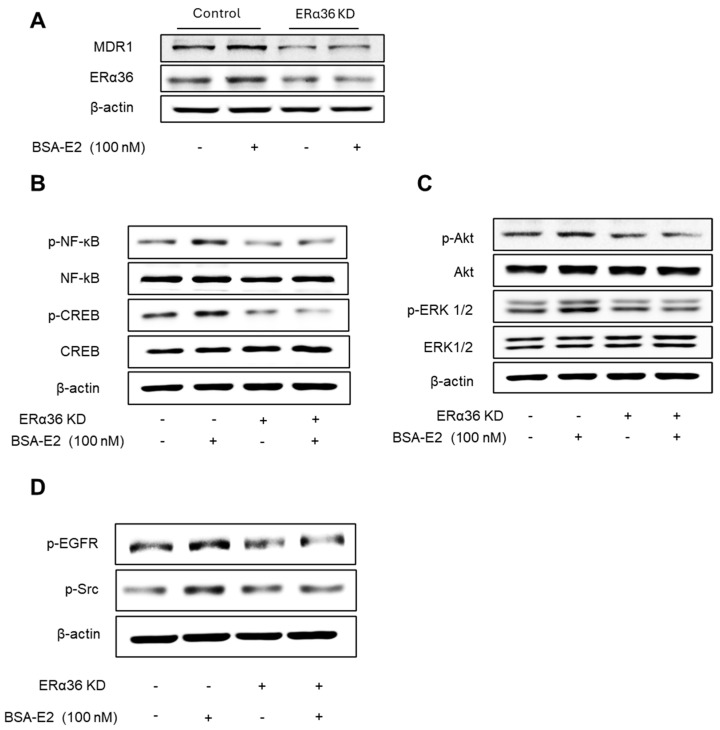 Figure 7
Figure 7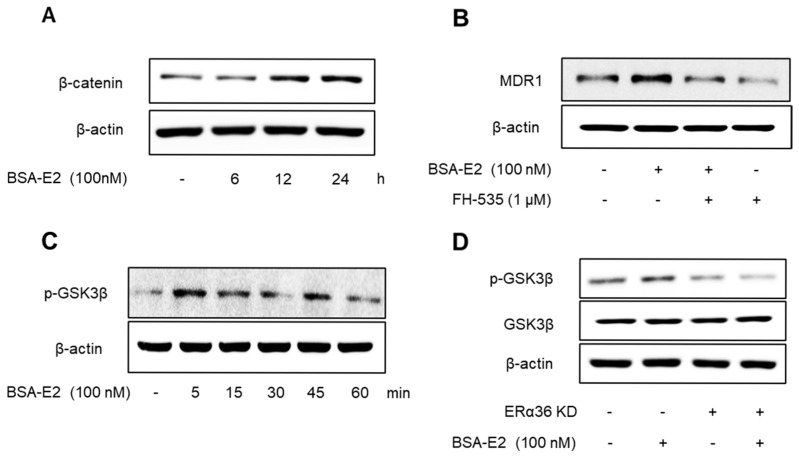 Figure 8
Figure 8- —National Research Foundation of Korea (NRF)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEstrogen and related hormone effects · Drug Transport and Resistance Mechanisms · NF-κB Signaling Pathways
1. Introduction
Breast cancer presents as a global disease burden, ranking as the most prevalent cancer in women and the second most frequently diagnosed cancer overall [1]. Despite significant advancements in early detection and therapeutic strategies, treatment resistance remains a critical challenge in clinical oncology. Among various breast cancer subtypes, triple-negative breast cancer (TNBC) accounts for approximately 15–20% of all breast cancer cases and is mainly observed in younger women [2]. TNBC is defined by the lack of estrogen receptors (particularly estrogen receptor 66 alpha, ERα66), progesterone receptors (PRs), and human epidermal growth factor receptor 2 (HER2) expression in tumor cells. This absence of conventional therapeutic targets classified TNBC as receptor-negative, contributing to its poor prognosis and limited treatment options [3,4]. In clinical practice, chemotherapy remains the mainstay of systemic treatment for TNBC because it does not respond to endocrine therapies or HER2-targeted agents. Unfortunately, TNBC often develops multidrug resistance (MDR) and shows poor long-term outcomes [5].
The majority of breast tissue is hormone-dependent, especially on estradiol, and is controlled by estrogen secretion. Given this dependency, estrogen signaling is critically involved in the development and metastasis of breast cancer through genomic and non-genomic signaling pathways [6,7]. This signaling is mediated by estrogen receptors (ERs), primarily Erα and Erβ. These receptors are ligand-activated transcription factors that, upon binding to 17β-estradiol (E2)—the predominant estrogen in the body—form homo- or heterodimers (E2-ER complexes). These complexes then translocate to the nucleus to initiate the transcription of target genes via the genomic pathway [8]. In parallel, a rapid non-genomic pathway can be triggered by E2 binding to ERs located at the membrane or in the cytoplasm. Upon binding, it triggers kinase cascade activation and intracellular calcium signaling such as phosphatidylinositide3-kinase/protein kinase B (PI3K/Akt), mitogen-activated protein kinase/extracellular regulated kinase (MAPK/ERK), and cAMP/PKA pathways [9]. These activations influence multiple cellular processes like proliferation, migration, and survival [9,10,11].
In recent years, research attention has turned to alternative isoforms of ERs as potential targets for treating TNBC, which lacks the classical ERα66. Among ER isoforms, ERα36 has gained attention due to its distinct localization and function. ERα36 is a 36 kDa splice variant of ERα66 that lacks both transactivation domains AF-1 and AF-2 and possesses distinct N- and C-termini [12,13]. Unlike ERα66, ERα36 is primarily found at the plasma membrane and in the cytoplasm, where it mediates a rapid membrane-initiated non-genomic estrogen signaling pathway [14,15]. Through this pathway, ERα36 is linked to the imitation of MAPK and PI3K/Akt signaling cascades. Previous studies have demonstrated that MAPK and Akt activation are correlated with cell growth and proliferation as well as the invasion and metastasis of various cancers [16,17,18,19]. Notably, ERα36 is frequently co-expressed with membrane proteins, such as the epidermal growth factor receptor (EGFR), in TNBC cells. This co-expression contributes to tumor growth through the formation of proto-oncogene tyrosine protein kinase (Src)/EGFR signaling complexes [20,21,22]. Elevated ERα36 expression is particularly observed in ER-negative, TNBC, and HER2-positive breast cancers [20,23,24]. In TNBC tumors, ERα36 is correlated with drug resistance, suboptimal responses to chemotherapy, and the promotion of epithelial–mesenchymal transition (EMT). Clinical data indicate that patients whose tumors lack ERα36 expression respond more favorably to chemotherapy than those with ERα36-positive tumors [25]. Chaudhri et al. demonstrated that ERα36 enhances the proliferation of breast cancer cells by regulating the expression of adhesion molecules and proteins involved in EMT [26].
Multidrug resistance plays a critical role in limiting the efficacy of breast cancer treatment and contributes substantially to therapeutic failure. A key mechanism underlying MDR involves ATP-binding cassette (ABC) family transporters, also known as multidrug resistance transporters, which actively mediate drug efflux in cancer cells [27]. The ATP-dependent pumps enable cancer cells to evade cytotoxic effects by limiting intracellular drug accumulation. Alternative model suggests that P-glycoprotein (P-gp), encoded by the MDR1 gene, may function as a flippase, transporting substrates from the cytosolic side to the external leaflet of the lipid bilayer [28,29]. Elevated MDR1 (P-gp) expression promotes the active efflux of various hydrophobic anticancer drugs, ultimately reducing the efficacy of chemotherapy [30]. The transcription of MDR1 is tightly regulated by various transcription factors. Among these, nuclear factor-κB (NF-κB), cAMP response elements (CREB), and β-catenin have been shown to bind to regulatory elements within the MDR1 promoter and enhance its transcription [31]. These factors are known to be activated by membrane-initiated signaling pathways. For example, the activation of NF-κB through pathways involving Src kinase, PI3K/Akt, or MAPKs leads to IκB phosphorylation and degradation, which enables NF-κB nuclear translocation and the subsequent activation of genes involved in inflammation, survival, and drug resistance [32,33]. MDR1 activation was reported to occur through NF-κB activation, linking inflammatory and survival signaling to drug resistance in cancer cells [34,35]. Emerging evidence indicates that ER signaling and the Wnt/β-catenin pathways converge to enhance MDR1 expression. Both ERα and β-catenin can directly activate MDR1 transcription via binding to its promoter region. In MCF7 breast cancer cells, the interaction of ERα with WW domain-binding protein 2 (WBP2) actively modulates MDR1 expression and contributes to doxorubicin resistance [36]. Meanwhile, the Wnt/β-catenin signal is triggered in doxorubicin-induced MDR cancer cells, where β-catenin associates with the CREB binding protein (CBP) to drive MDR1 expression in an MEK/ERK-dependent manner [37]. While biologically relevant crosstalk between ER and Wnt/β-catenin signaling has been implicated in fostering chemoresistance, the direct molecular synergy between these pathways in the regulation of MDR1 has not been fully evaluated.
Although ERα36 is important for the tumorigenesis and progression in breast cancer, the specific mechanisms through which it contributes to MDR1 expression in TNBC remain poorly understood. Addressing this gap, we aimed to elucidate the role of ERα36 in regulating MDR1 expression in MDA-MB-231 breast cancer cells, with a particular focus on its potential crosstalk with the Wnt/β-catenin pathway. Delineating this relationship may provide insights into overcoming drug resistance in aggressive cancer subtypes.
2. Results
2.1. BSA-E2 Modulates Adriamycin Resistance by Enhancing MDR1-Mediated Efflux
Adriamycin (also known as doxorubicin) is a widely used chemotherapeutic agent, especially for treating breast cancer, leukemia, lymphomas, and sarcomas. However, Adriamycin resistance poses a major obstacle to effective cancer therapy. In ER-positive tumors, estrogen signaling is known to influence drug response. To specifically assess the involvement of membrane estrogen receptor signaling in Adriamycin-induced cytotoxicity, we employed BSA-conjugated estradiol (BSA-E2), which is membrane-impermeable and thus selectively activates membrane-associated estrogen receptors without triggering the genomic signaling pathway. By assessing cell viability following co-treatment with Adriamycin and BSA-E2, we realized that BSA-E2 increased the cell viability of MDA-MB-231 cells while it attenuated the cytotoxicity caused by Adriamycin (Figure 1A,B). Because Adriamycin-induced drug resistance is often associated with increased MDR1 expression, we further investigated whether treatment with BSA-E2 suppresses the expression of MDR1 using Rhodamine-123 (Rh-123) accumulation assays. Figure 1C shows that treatment with verapamil, an MDR1 inhibitor, efficiently increased the accumulation of the Rh123 fluorescent signal, indicating that MDR1 is associated with drug resistance. Cells treated with BSA-E2 showed a notable reduction in intracellular Rh-123 levels, suggesting that BSA-E2 likely increases MDR1 activity or expression, thereby promoting drug efflux and potentially contributing to drug resistance.
2.2. BSA-E2 Increases MDR1 Expression in MDA-MB-231 Cells
Since the development of the MDR1 phenotype in cancer cells is linked to MDR1 overexpression, we further examined the change in MDR1 mRNA levels upon treatment with BSA-E2 using RT-PCR. BSA-E2 treatment significantly increased the mRNA level of MDR1 in a time- and concentration-dependent manner (Figure 2A,B). Together, Western blot exhibited an elevated protein level of MDR1 (Figure 2C,D; quantified in Supplementary Figure S1A,B). Taken together, these data suggest that BSA-E2 promotes MDR1 expression in MDA-MB-231 cells.
2.3. BSA-E2 Induces MDR1 Expression via Activation of NF-κB and CREB Phosphorylation
MDR1 expression is governed by a variety of transcription factors, including NF-κB, particularly the p65 (RelA) subunit, and CREB, in which CREB binds to the cAMP response element to modulate the expression of MDR1 in breast cancer cells [38,39]. To investigate whether CREB and NF-κB activation are involved in Erα36-mediated MDR1 expression in MDA-MB-231 cells, we examined the activation of CREB and NF-κB in response to BSA-E2 treatment. BSA-E2 significantly increased CREB and NF-κB phosphorylation in a time- and concentration-dependent manner (Figure 3A,B; quantified in Supplementary Figure S2A,B). To further clarify the effects of BSA-E2 on NF-κB and CREB in BSA-E2-induced MDR1 expression, specific inhibitors, Bay 11-7082 (an NF-κB inhibitor) and 666-15 (a CREB inhibitor), were used. Pretreatment with Bay-117082 and 666-15 suppressed BSA-E2-induced MDR1 expression (Figure 3C,D; quantified in Supplementary Figure S2C,D). These results suggest that the transcriptional activity of NF-κB and CREB is important for BSA-E2-mediated regulation of MDR1 in MDA-MB-231 cells.
2.4. Akt/ERK Signaling Pathway Is Important for ERα36-Mediated MDR1 Expression
Given that NF-κB and CREB are regulated through the Akt and ERK signaling pathways, we next investigated whether BSA-E2 influences the activation of these upstream pathways. Cells treated with BSA-E2 exhibited an increase in the phosphorylation of Akt and ERK in a manner dependent on both time and concentration (Figure 4A,B; quantified in Supplementary Figure S3A,B). Interestingly, pretreatment of LY294002 (a PI3K/Akt inhibitor) and PD98059 (an ERK inhibitor) inhibited CREB and NF-κB phosphorylation as well as the expression of MDR1 (Figure 4C,D; quantified in Supplementary Figure S3C,D). These findings indicate that Akt and ERK activity are crucial for MDR1 upregulation by BSA-E2.
2.5. BSA-E2 Activates EGFR/Src-Akt/ERK Signaling to Induce MDR1 Expression
Following the observed activation of Akt and ERK, we explored whether EGFR and Src—known upstream regulators of these pathways—are also activated in response to BSA-E2. As shown in Figure 5A,B, BSA-E2 induced the phosphorylation of EGFR and Src in MDA-MB-231 cells (the band density was quantified in Supplementary Figure S4A,B). Pretreatment with AG1478 (an EGFR inhibitor) or PP2 (a Src inhibitor) attenuated the BSA-E2-induced expression of MDR1 and the phosphorylation of Akt, ERK, CREB, and NF-κB (Figure 5C,D; quantified in Supplementary Figure S5C,D), suggesting that the activation of EGFR and Src plays a key upstream role in regulating MDR1 expression.
2.6. ERα36 Mediates BSA-E2-Induced Drug Resistance and Cell Survival in MDA-MB-231 Cells
Given the established role of membrane-associated signaling in BSA-E2-induced MDR1 expression, we next investigated whether ERα36 contributes to this regulation by monitoring the cell viability and cytotoxicity in the ERα36 knocked down cells in response to Adriamycin. ERα36 knocked down cells (ERα36 KD) exhibited a notable decrease in cytotoxicity and viability compared with wild-type cells (Control) (Figure 6A). To further investigate the role of ERα36 in BSA-E2-induced drug resistance, we assessed the cell viability and cytotoxicity following treatment with 100 nM of BSA-E2. In ERα36-knockdown cells, BSA-E2 had no effect; however, in wild-type cells, BSA-E2 treatment led to a marked reduction in the cytotoxicity and a corresponding increase in cell viability (Figure 6B,C). Additionally, the downregulation of ERα36 influenced the expression of multidrug resistance genes and proteins, as indicated by Rh-123 accumulation. Treatment with BSA-E2 did not alter the intracellular level of Rh-123 in ERα36 KD cells, whereas a decrease was observed in wild-type cells, suggesting that ERα36 is required for BSA-E2-induced drug efflux activity, likely through the upregulation of multidrug resistance proteins. Collectively, these findings indicate that ERα36 contributes to drug resistance and cell survival in MDA-MB-231 cells.
2.7. ERα36 Is Important for Mediating BSA-E2-Induced MDR1 Expression Through Akt/ERK and NF-κB/CREB Signaling Pathway
We next explored the role of ERα36 in regulating signaling pathways involved in BSA-E2-induced MDR1 expression. ERα36 KD cells exhibited a markedly lower level of the MRD1 protein compared with wild-type cells (Figure 7A; quantified in Supplementary Figure S5A). Notably, BSA-E2 treatment failed to induce MRD1 expression in ERα36 KD cells, in contrast to the response observed in control cells. In addition, the ERα36 knockdown suppressed BSA-E2-induced phosphorylation of NF-κB, CREB, Akt, EGFR, and Src (Figure 7B–D; quantified in Supplementary Figure S5B–D). These results demonstrate that ERα36 is located on the plasma membrane and mediates BSA-E2-stimulated Akt/ERK activation through the Src/EGFR signaling pathway, resulting in increased MDR1 expression.
2.8. ERα36 Mediates BSA-E2-Induced Activation of Wnt/β-Catenin Signaling and MDR1 Expression
Emerging evidence suggests that the NF-κB/CREB signaling pathways interact functionally with the Wnt/β-catenin pathway to modulate gene expression. Therefore, we investigated whether ERα36 mediates its downstream effects through the activation of the Wnt/β-catenin signaling pathway. Treatment with BSA-E2 significantly increased the β-catenin levels (Figure 8A; quantified in Supplementary Figure S6A), while pretreatment with FH-535, a Wnt/β-catenin inhibitor, downregulated the expression of MDR1 (Figure 8B and Figure S6B), suggesting that Wnt/β-catenin signaling plays a role in the regulation of MDR1. Notably, BSA-E2 treatment altered GSK3β phosphorylation, with a marked increase observed at 5 min post-treatment (Figure 8C and Figure S6C). This phosphorylation was abolished upon ERα36 knockdown (Figure 8D and Figure S6D). These data together reveal that the BSA-E2 activates the Wnt/β-catenin signaling pathway through an ERα36-dependent mechanism.
3. Discussion
P-glycoprotein, the product of the MDR1 gene, has received great interest for its contribution to multidrug resistance in numerous cancer types [40]. The overexpression of MDR1 is a major obstacle to effective cancer chemotherapy. Moreover, its role in enabling cancer cells to evade chemotherapy-induced apoptosis has been demonstrated in several cellular models, particularly in breast cancer cells, including TNBC [40,41]. TNBC characterized by a high expression of ERα36 has increased drug resistance, which leads to cell proliferation, metastasis, and malignancy, suggesting the potential significance of ERα36 status in predicting patient response to chemotherapy [25]. In this study, we provide evidence that ERα36 contributes to the acquisition of chemoresistance in TNBC by upregulating MDR1 expression through a non-genomic pathway.
The activation of ERα36 by BSA-E2 significantly inhibited Adriamycin-induced cytotoxicity and markedly increased MDR1 expression. Furthermore, treatment with BSA-E2 triggered the MDR1-dependent drug efflux evaluated by the reduced accumulation of its substrate, Rh-123. On the other hand, the silencing of ERα36 attenuated BSA-E2-induced MDR1 expression and no longer exhibited effects on the suppression of Adriamycin-induced lethality. This suggests the contribution of ERα36 to estrogen-mediated drug resistance through the upregulation of MDR1. In the context of MDR1 regulation, we explored the involvement of ERα36 in multiple interconnected signaling pathways.
3.1. ERα36-Akt/ERK/CREB and NF-κB Pathway
MDR1 transcription is under the control of multiple transcription factors, such as NF-κB, CRE, AP-1, SP1, and PXR [42,43]. Our data demonstrate that BSA-E2 activates multiple non-genomic signaling pathways, notably the Akt/ERK/CREB and NF-κB axes. The inhibition of Akt or ERK suppressed BSA-E2-induced MDR1 expression. Moreover, these effects were significantly attenuated upon the knockdown of ERα36, indicating that this receptor mediates the upstream activation of these kinases. The Akt and ERK pathways are well-established regulators of cell survival and drug resistance [19,44]. Furthermore, MDR1 transcription is directly regulated by CREB and NF-κB through their binding to specific sites in the distal promoter region of the MDR1 gene [35]. Phosphorylated CREB binds to its promoter region and enhances MDR1 transcription. Similarly, NF-κB, especially the p65 (RelA) subunit, plays a critical role in MDR1 regulation. Upon activation by cellular stress or chemotherapeutic agents, p65 translocates to the nucleus and binds to a consensus κB motif in the first intron of the MDR1 gene, promoting its transcriptional activation [35,45]. This mechanism contributes to decreased intracellular drug accumulation and treatment failure in various cancer types. These results collectively support a model in which ERα36 activates the Akt/ERK signaling to drive MDR1 regulation, contributing to enhanced drug resistance.
3.2. ERα36-EGFR/Src Signaling
We observed that BSA-E2 treatment rapidly induces phosphorylation of both EGFR and Src, whereas this signal is altered in ERα36 knockdown, suggesting a cross-correlation of action between EGFR and ERα36. ERα36 activates membrane-mediated estrogen signaling in association with EGFR and Src, resulting in the regulation of MDR1 expression through the Akt and ERK signaling pathways. Indeed, a linkage between EGFR and ERα36 expression in carcinoma cells was determined, indicating that ERα36 influences the activation of extracellular signaling involved in EGFR/Src [16,44], probably through the interaction with membrane proteins of breast cancer cells. In particular, Src has been shown to serve as a key intermediary in this process by facilitating EGFR phosphorylation in response to estrogenic stimuli. Our data support this mechanism, implicating EGFR and Src as early effectors in the ERα36 signaling axis, ultimately contributing to MDR1 regulation.
3.3. ERα36-Wnt/β-Catenin Pathway
Beyond classical kinase signaling, our data implicate the Wnt/β-catenin pathway as a downstream effector of ERα36 in promoting MDR1 expression. The enhanced β-catenin protein levels and phosphorylation of GSK3β suggest that ERα36 plays roles in the activation of canonical Wnt signaling, which has been widely linked to tumor progression and multidrug resistance. It has been reported that the activation of the Wnt/β-catenin pathway enhances ABC transporter transcription, thereby contributing to chemoresistance across multiple cancer types [37,46]. In particular, the β-catenin/T-cell factor 4 transcriptional complex has been shown to directly target the MDR1 gene and increase MDR1 expression [47]. Furthermore, consistent with previous studies [48,49], our data demonstrated that the transactivation of MDR1 can be inhibited by the pharmacological suppression of upstream regulators. Specifically, the inhibition of Src by PP2 and β-catenin by FH535 or the silencing of ERα36 effectively prevents MDR1 expression, suggesting functional crosstalk between ER signaling and Wnt/β-catenin signaling pathways. Collectively, our data suggest that ERα36 simultaneously engages kinase cascades and developmental pathways like Wnt/β-catenin to orchestrate a coordinated response that enhances drug efflux and promotes resistance.
Taken together, our study reveals that ERα36 serves as a central hub in coordinating non-genomic estrogen signaling, promoting MDR1 expression and resistance to Adriamycin. These effects are mediated through the activation of multiple pathways—Akt/ERK/CREB, NF-κB, and Wnt/β-catenin—as well as through interactions with EGFR. Importantly, the knockdown of ERα36 abolished most of these downstream events, confirming its essential role in this network. Our finding provides molecular evidence that a single receptor, like ERα36, can orchestrate diverse downstream oncogenic pathways. This highlights its potential as a therapeutic target to simultaneously overcome various mechanisms of drug resistance during treatment. Although our study focuses on breast cancer cells, particularly TNBC, the role of ERα36 may extend beyond this context. Previous studies have reported ERα36 expression in other cancer types, including lung, gastric, and endometrial cancers, suggesting its broader involvement in tumor biology. Future comparative studies are necessary to determine the generalizability of this mechanism. In addition, validation using animal models would be critical to investigate how ERα36 knockdown influences key tumor behaviors within the complex tumor microenvironments. Models such as patient-derived xenograft (PDX) or orthotopic mouse models could offer physiologically relevant settings that closely mimic patient responses to chemotherapy [50]. Furthermore, innovative drug delivery platforms, such as nanoparticle-based therapeutics, represent promising strategies to effectively target ERα36 in resistant cancer types [51].
These findings not only elucidate a new mechanism of estrogen-induced multidrug resistance in breast cancer but also point to ERα36 as a potential therapeutic target, particularly in tumors that are ERα-negative but still responsive to estrogen at the membrane level. Future studies may focus on identifying specific inhibitors of ERα36 or targeting its associated pathways to overcome resistance and improve chemotherapeutic efficacy.
4. Materials and Methods
4.1. Chemicals and Reagents
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin–streptomycin, and trypsin were purchased from Welgene (Gyeongsan, South Korea). Antibodies against ERα36 were purchased from Alpha Diagnostic International Inc. (San Antonio, TX, USA). BSA-conjugated estradiol (BSA-E2) was obtained from Sigma-Aldrich (St. Louis, MO, USA). The following primary antibodies from Cell Signaling Technology (Danvers, MA, USA) were used at a 1:1000 dilution: MDR1 (Cat#13342S), p-EGFR (Tyr1068 specific, Cat#2234S), EGFR (Cat#2232L), p-c-Src (Tyr416 specific, Cat#2101S), p-Akt (Ser472 specific, Cat#9271), Akt (Cat#9272S), p-ERK (Thr202/Tyr204 specific, Cat#9101), ERK (Cat#9102), p-NF-κB p65 (Ser536 specific, Cat#3033)), NF-κB p65 (Cat#8242), CREB (Cat#4820), p-CREB (Ser133 specific, Cat#9198). HRP-conjugated anti-rabbit (Cat#sc-2357) or anti-mouse (Cat#sc-2005) secondary antibodies from Santa Cruz Biotechnology (Dallas, TX, USA) were used at a 1:5000 dilution. Verapamil, rhodamine, and Adriamycin were purchased from the Sigma Chemical Company (St. Louis, MO, USA). RNAiso was purchased from Takara Bio (Shiga, Japan). Antibody against β-actin was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a tetrazole) (MTT) was purchased from USB Corporation (Cleveland, OH). All other commercially available chemicals used were of the highest purity.
4.2. Cell Culture and Cell Viability Assays
The human breast cancer cell line MDA-MB-231 was provided by the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were grown in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL of penicillin, and 100 μg/mL streptomycin at 37 °C in an atmosphere containing 5% CO_2_. To assess cell viability, cells were plated in 48-well plates at 2 × 10^4^ cells/well, and 100 nM of BSA-E2 was added to each well after 24 h incubation. The MTT and LDH assays were performed as described previously [52]. Briefly, cells were treated with MTT for 1 h, and formazan crystals were solubilized with DMSO. Absorbance was measured at 570 nm with a Biotek Synergy HT microplate reader (BioTek Instruments, Winooski, VT, USA). LDH activity in the supernatant was determined at 490 nm.
4.3. Lentiviral Vector Production and Transduction
Lentiviral ERα36 shRNA vectors (HSH859L-1-LVRU6GP) for ERα36 knockdown and lentiviral control (CSHCTR00-LVRU6GP) vectors were purchased from Genecopoeia (Rockville, MD, USA). Following the manufacturer’s instructions, all lentiviral titers were produced using a Lenti-PacTM FIV Expression Packaging Kit (Genecopoeia, MD, USA). ERα36-knockdown stable cell lines were generated by transducing MDA-MB-231 cells with purified virus, followed by a selection of stable pools of cells using 5 μg/mL of puromycin (Sigma-Aldrich, St. Louis, MO, USA).
4.4. RNA Extraction and qRT-PCR
Total RNA was extracted from harvested cell pellets containing approximately 1 × 10^6^ MDA-MB-231 cells using RNAiso Plus (total RNA extraction reagent; Takara Bio, Shiga, Japan). After RNA isolation, cDNA was synthesized using the BioFact RT Series kit (BioFact, Daejeon, Korea). The qRT-PCR results were analyzed using Bio-Rad CFX Connect Real-Time PCR software, version 1.4.1 (Bio-Rad Laboratories, Hercules, CA, USA). The PCR primers were used as follows. The expression was normalized with the endogenous control, glyceraldehyde 3-phosphate dehydrogenase (GAPDH). MDR1 (NM_001348945.2) forward: 5′-GCTGTCAAGGAAGCCAATGCCT-3′, MDR1 (NM_001348945.2) reverse: 5′-TGCAATGGCGATCCTCTGCTTC-3′; GAPDH (NM_001256799.3) forward: 5′-GTCTCCTCTGACTTCAACAGCG-3′, GAPDH (NM_001256799.3) reverse: 5′-ACCACCCTGTTGCTGTAGCCAA-3′.
4.5. Western Blotting
Western blotting was performed according to the standard protocol [31]. Briefly, approximately 1 × 10^7^ cells were harvested and lysed using CETi lysis buffer (TransLab, Daejeon, Korea). An equal amount of total protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane. The membranes were blocked using skim milk for 1 h. Subsequently, primary antibodies were incubated at 4 °C overnight, and secondary antibodies were incubated at RT for 2 h. The membrane was exposed using the Hisol ECL Plus detection kit (Biofact, Daejeon, Korea).
4.6. Rhodamine-123 Accumulation Assay
Cells were plated onto 24-well plates (at 10^5^ cells/well) and pretreated with 10−100 nM of BSA-E2 and 20 μM of verapamil for 48 hr. Verapamil was used as a positive control for MDR inhibition [53]. Following pretreatment, cells were cultured in medium containing 5 μM of Rh-123 for 90 min, protected from light. Cells were trypsinized, washed twice with ice-cold PBS, and resuspended in 1 mL of PBS. Intracellular Rh123 accumulation was measured by fluorescence at 488 nm excitation and 530 nm emission using a BioTek Synergy HT microplate reader (BioTek Instruments, Winooski, VT, USA).
4.7. Statistical Analysis
All experiments were performed in triplicate. The data are reported as mean ± SD of independent experiments. The Shapiro–Wilk test was used for data normality. The statistical evaluation of the results was performed using one-way ANOVA. The Tukey–Kramer test was used for multi-group comparisons. Statistical significance was defined as p < 0.01.
5. Conclusions
In summary, this study demonstrates that ERα36 contributes to drug resistance in TNBC cells by promoting MDR1 expression through multiple non-genomic pathways, including Akt/ERK, NF-κB, CREB, and Wnt/β-catenin. These findings expand the functional repertoire of ERα36 beyond tumor growth and suggest its involvement in regulating key resistance mechanisms, positioning it as a potential therapeutic target in drug-resistant breast cancers.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kolak A. Kamińska M. Sygit K. Budny A. Surdyka D. Kukiełka-Budny B. Burdan F. Primary and secondary prevention of breast cancer Ann. Agric. Environ. Med.20172454955310.26444/aaem/7594329284222 · doi ↗ · pubmed ↗
- 2Carey L.A. Perou C.M. Livasy C.A. Dressler L.G. Cowan D. Conway K. Karaca G. Troester M.A. Tse C.K. Edmiston S. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study JAMA 20062952492250210.1001/jama.295.21.249216757721 · doi ↗ · pubmed ↗
- 3Bianchini G. Balko J.M. Mayer I.A. Sanders M.E. Gianni L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease Nat. Rev. Clin. Oncol.20161367469010.1038/nrclinonc.2016.6627184417 PMC 5461122 · doi ↗ · pubmed ↗
- 4Foulkes W.D. Smith I.E. Reis-Filho J.S. Triple-negative breast cancer N. Engl. J. Med.20103631938194810.1056/NEJ Mra 100138921067385 · doi ↗ · pubmed ↗
- 5Dent R. Trudeau M. Pritchard K.I. Hanna W.M. Kahn H.K. Sawka C.A. Lickley L.A. Rawlinson E. Sun P. Narod S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence Clin. Cancer Res.2007134429443410.1158/1078-0432.CCR-06-304517671126 · doi ↗ · pubmed ↗
- 6Pedram A. Razandi M. Lewis M. Hammes S. Levin E.R. Membrane-localized estrogen receptor α is required for normal organ development and function Dev. Cell 20142948249010.1016/j.devcel.2014.04.01624871949 PMC 4062189 · doi ↗ · pubmed ↗
- 7Pedram A. Razandi M. Blumberg B. Levin E.R. Membrane and nuclear estrogen receptor α collaborate to suppress adipogenesis but not triglyceride content FASEB J.20163023024010.1096/fj.15-27487826373802 PMC 4684544 · doi ↗ · pubmed ↗
- 8Björnström L. Sjöberg M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes Mol. Endocrinol.20051983384210.1210/me.2004-048615695368 · doi ↗ · pubmed ↗