Comparative Transcriptomics and Metabolomics Uncover the Molecular Basis of Leaf Rust Resistance in Contrasting Leymus chinensis Germplasms
Wenxin Gao, Peng Gao, Fenghui Guo, Xiangyang Hou

TL;DR
This study uses transcriptomics and metabolomics to uncover the molecular mechanisms behind rust resistance in a forage grass called Leymus chinensis.
Contribution
The study identifies key genes and metabolites associated with rust resistance in Leymus chinensis using multi-omics integration.
Findings
1012 differentially expressed genes were identified, with notable changes in cell wall biosynthesis and photosynthesis pathways.
287 differentially accumulated metabolites were found, including suppressed flavonoids and upregulated cutin synthesis in resistant germplasms.
79 co-enriched pathways were uncovered, highlighting nucleotide metabolism and flavonoid biosynthesis as critical for rust resistance.
Abstract
Leymus chinensis (Trin.) Tzvel., a vital native forage grass in northern China for ecological restoration and livestock production, faces severe yield losses and grassland degradation due to rust (Puccinia spp.) infection. Current control strategies, reliant on chemical interventions, are limited by evolving resistance risks and environmental concerns, while rust-resistant breeding remains hindered by insufficient molecular insights. To address this, we systematically evaluated rust resistance in 24 L. chinensis germplasms from diverse geographic origins, identifying six highly resistant (HR) and five extremely susceptible (ES) genotypes. Integrating transcriptomics and metabolomics, we dissected molecular responses to Puccinia infection, focusing on contrasting HR (Lc71) and ES (Lc5) germplasms at 48 h post-inoculation. Transcriptomic analysis revealed 1012 differentially expressed…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1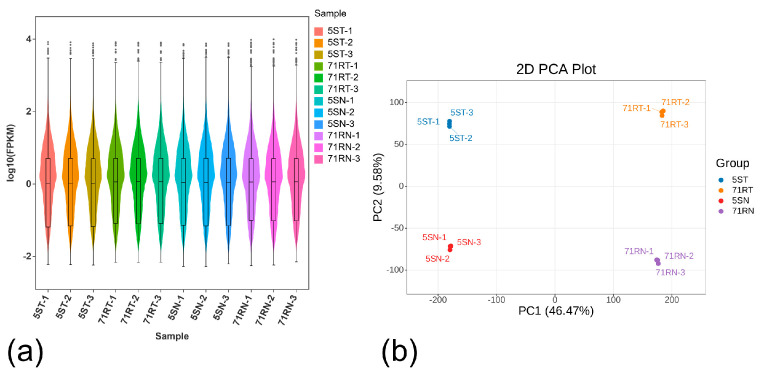 Figure 2
Figure 2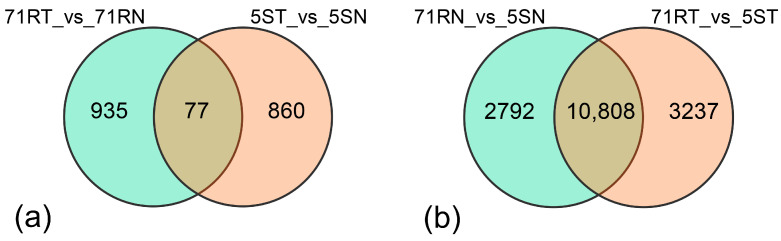 Figure 3
Figure 3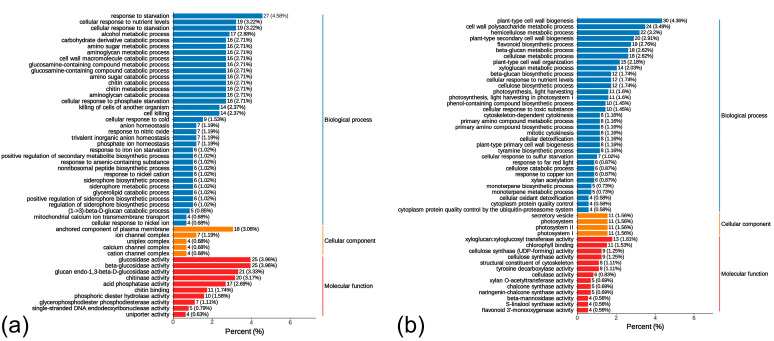 Figure 4
Figure 4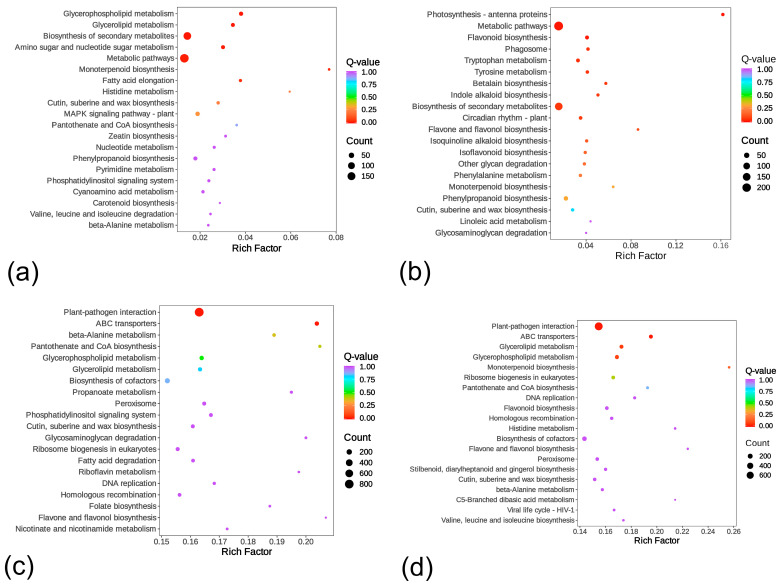 Figure 5
Figure 5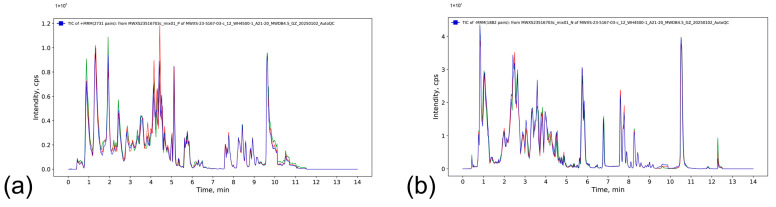 Figure 6
Figure 6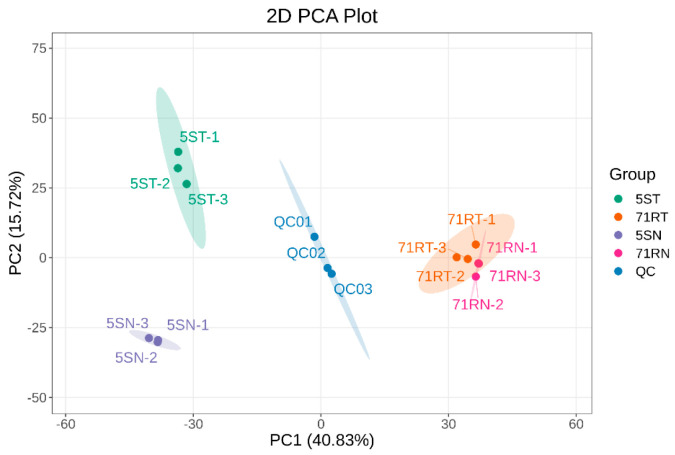 Figure 7
Figure 7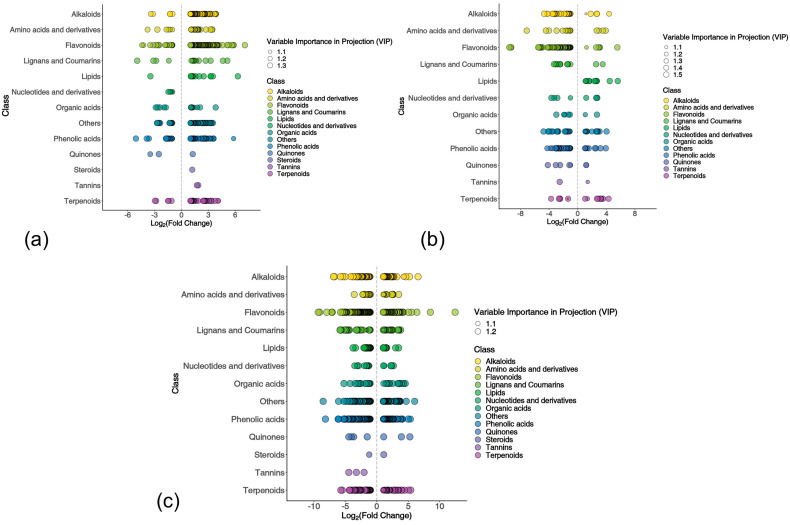 Figure 8
Figure 8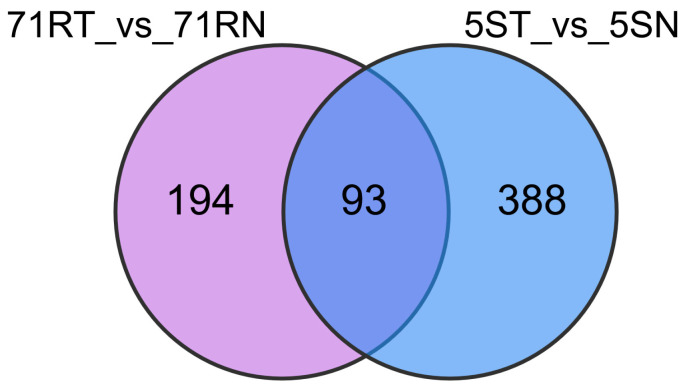 Figure 9
Figure 9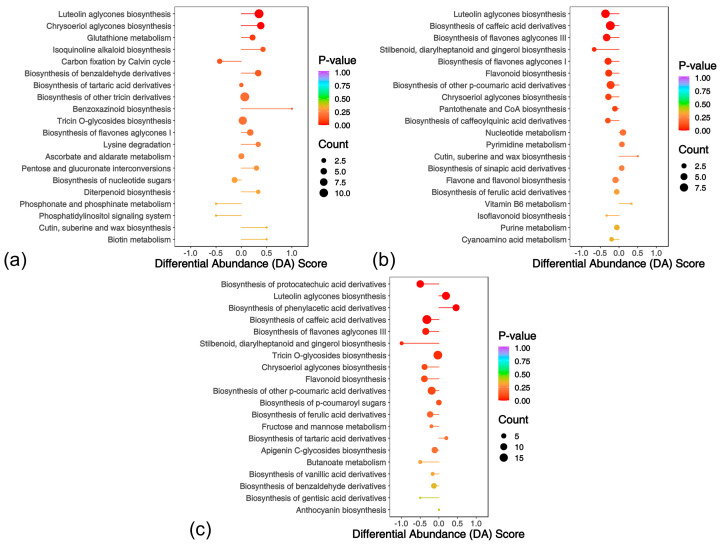 Figure 10
Figure 10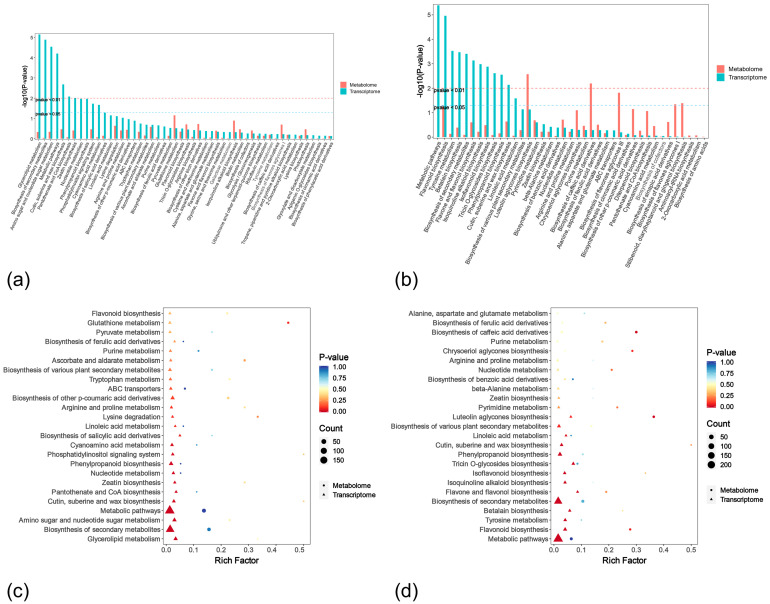 Figure 11
Figure 11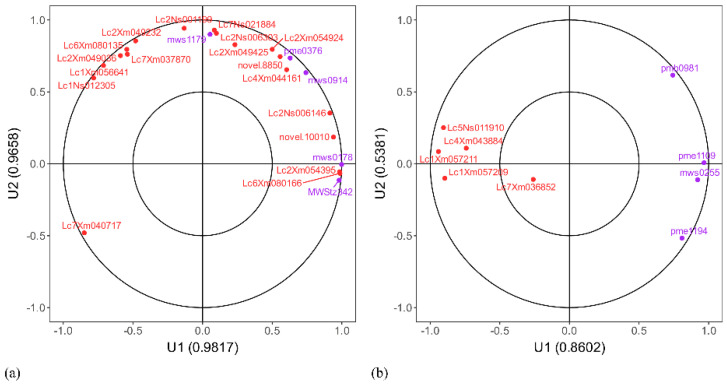 Figure 12
Figure 12- —Shanxi Provincial Key Research and Development Program (Agriculture Field)
- —Fundamental Research Program of Shanxi Province
- —Shanxi Youth Science Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant-Microbe Interactions and Immunity · Genetic and Environmental Crop Studies · Medicinal Plant Research
1. Introduction
Leymus chinensis (Trin.) Tzvel., designated as alkali grass, is a perennial rhizomatous forage species within the genus Leymus (Poaceae family) [1]. This species dominates Eurasian steppe ecosystems, with its core distribution spanning northeastern China, Inner Mongolia, Hebei, and Shanxi provinces, where it occupies >50% of temperate grasslands [2,3]. As a premium native forage in China, L. chinensis has high biomass yield and nutritional richness, with excellent palatability [4]. Moreover, its extensive root system confers multipronged stress tolerance, including robust cold hardiness, drought resistance, and saline–alkaline adaptation [5], underpinning critical roles in restoring degraded grasslands and combating desertification [6].
However, the ecological functions of L. chinensis are under significant threat from rust disease. Rust is a common fungal disease in Poaceae grasslands [7]. Pathogenesis primarily targets leaves, sheaths, and stems [8], causing yield losses, quality deterioration, and plant mortality under severe infection [9,10]. Currently, the control of grass rust diseases mainly relies on agricultural practices such as proper planting density, timely pruning, prompt removal of diseased plant residues, and chemical control using fungicides like triadimefon, propiconazole, and myclobutanil [11,12,13], yet faces three inherent constraints: chemical applications risk for environmental contamination, pathogen resistance evolution, and human/livestock toxicity; cultural methods exhibit limited efficacy and cost-effectiveness; both approaches prove impractical for vast natural grasslands with complex ecosystems [14,15]. Therefore, considering environmental friendliness and safety, actively screening, breeding, and utilizing rust-resistant varieties of L. chinensis is one of the most economical and effective measures for controlling rust disease in L. chinensis [9,16]. The collection of germplasm resources and evaluation of disease resistance are prerequisites for the screening of disease-resistant germplasm resources and breeding of disease-resistant varieties [17].
Plants deploy a layered immune system against pathogens [18,19]. The primary barrier, pattern-triggered immunity (PTI), is initiated when cell-surface pattern recognition receptors (PRRs) detect pathogen-associated molecular patterns (PAMPs) [20,21,22,23], activating basal defenses including reactive oxygen species (ROS) bursts and cell wall fortification. The secondary tier, effector-triggered immunity (ETI), engages when pathogenic effectors suppress PTI; intracellular nucleotide-binding leucine-rich repeat (NLR) proteins then recognize these effectors [24,25], eliciting amplified responses such as hypersensitive response highly resistant (HR) and systemic acquired resistance (SAR) [26,27,28]. Moreover, PTI and ETI do not operate as fully discrete defense tiers. Emerging evidence indicates that ETI activation requires the involvement of PRRs and their co-receptors, while ETI-derived signaling modulates PTI pathways, thereby amplifying PTI responses [29,30].
Recent omics advancements provide unprecedented resolution for dissecting disease resistance mechanisms [31]. Transcriptomics, exemplified by RNA sequencing (RNA-Seq), interrogates genome-wide expression dynamics via high-throughput sequencing [32], while metabolomics, employing liquid chromatography–mass spectrometry and gas chromatography–mass spectrometry (LC-MS/GC-MS), quantifies global metabolite flux [33]. Integration of these approaches overcomes inherent limitations of single-omics studies, elucidating molecular interactions between genes and metabolites during plant–pathogen responses [34,35,36].
L. chinensis, with its high nutritional value and stress resistance, has become an important species for ecological restoration and forage production in the northern grasslands of China [37,38]. However, rust disease reduces yield and causes degradation, severely restricting its production application and ecological function [39,40]. Currently, the breeding of rust-resistant L. chinensis faces bottlenecks such as unclear genetic background of germplasm resources, lack of resistance evaluation system, and unclear disease resistance molecular mechanisms [41]. Against this background, based on the previously established L. chinensis germplasm resource garden (covering wild germplasms from different geographical sources), this study accurately evaluated the rust resistance indoors, screened out germplasms with significantly differentiated resistance levels, and selected typical HR and ES germplasms to conduct joint analysis of transcriptomics and metabolomics. By dissecting the DEGs, DAMs, and their related pathways at 48 h post-inoculation with rust fungus, potential resistance regulatory genes were mined to provide theoretical support for the molecular breeding of rust-resistant L. chinensis.
2. Results
2.1. Identification and Evaluation of Rust Resistance in 24 Leymus chinensis Germplasms Under Laboratory Conditions
The resistance evaluation results revealed significant differences in the incidence and disease index (DI) among the 24 Leymus chinensis germplasms (p < 0.05, Table 1). Disease incidence ranged from 6.67% to 100%, with disease indices spanning 1.33–80.87. Based on DI grading, the germplasms were classified as follows: six highly resistant (HR) (25.00%; Lc30, Lc53, Lc62, Lc68, Lc70, Lc71), five moderately resistant (MR, 20.83%), three moderately susceptible (MS, 12.50%), five highly susceptible (HS, 20.83%), and five extremely susceptible (ES) (20.83%; Lc5, Lc19, Lc49, Lc55, Lc57). No immune germplasms were observed. Representative disease symptoms at 12 days post-inoculation are documented in Figure 1.
The significant differences in rust incidence and DI among these germplasms demonstrate diverse resistance levels. The HR germplasm Lc71 and ES germplasm Lc5 were selected for subsequent molecular analysis.
2.2. Transcriptomic Analysis of Resistant and Susceptible L. chinensis in Response to Rust Fungus Infection
2.2.1. RNA Sequencing and Quality Control of Samples
A total of 12 samples from Lc71 (HR) and Lc5 (ES) germplasms—encompassing both inoculated and non-inoculated treatments—underwent reference-based transcriptome sequencing. This yielded 98.79 Gb of Clean Data (high-quality filtered reads), with Q20 (percentage of bases with Phred quality score > 20) and Q30 (quality score >30) and guanine-cytosine (GC) content surpassing 53.42%. These metrics confirm high base-calling accuracy and sequencing integrity, ensuring data reliability for downstream analyses (Table S1).
2.2.2. Analysis of Gene Expression Levels and Principal Component Analysis (PCA) Between Samples
Figure 2a shows that the gene expression levels span six orders of magnitude across samples. Although expression distribution and probability density exhibit inter-sample similarity, discernible individual variations persist. Principal component analysis (PCA) of the 12-sample dataset (Figure 2b) reveals significant divergence between treatments of the two germplasms. Crucially, triplicate biological replicates under identical treatments exhibit clustered PC values, confirming minimal intra-group variation alongside substantial inter-group differences, thereby ensuring experimental reproducibility for subsequent analyses.
2.2.3. Screening of Differentially Expressed Genes (DEGs)
The volcano plot of differentially expressed genes DEGs (Figure S1) revealed significant transcriptional reprogramming in resistant and susceptible germplasms following Puccinia spp. infection. In the L. chinensis Lc5 (ES) treatment group (5ST vs. 5SN), 937 DEGs were identified, comprising 690 upregulated and 247 downregulated genes (Figure S1a). For the L. chinensis Lc71 (HR) treatment group (71RT vs. 71RN), 1012 DEGs were detected (247 upregulated and 765 downregulated (Figure S1b), suggesting broader stress-responsive transcriptional regulation in the resistant germplasm. Comparative analysis between genotypes identified 13,600 DEGs in 71RN vs. 5SN (resistant vs. susceptible control; 7201 upregulated), and 14,045 DEGs in 71RT vs. 5ST group (treatment comparison; 7309 upregulated; Figure S1c,d), indicating genotype-specific transcriptional regulation attributable to the resistance phenotype.
Further Venn diagram analysis (Figure 3) elucidated DEG interactions. Seventy-seven core DEGs were shared between L. chinensis Lc5 (ES) and L. chinensis Lc71 (HR) treatments, with 935 and 860 DEGs exclusive to resistant and susceptible germplasms, respectively (Figure 3a). Cross-group comparisons revealed 10,808 constitutive DEGs shared between 71RN vs. 5SN and 71RT vs. 5ST, alongside 2792 and 3237 treatment-specific DEGs (Figure 3b), demonstrating that resistance formation compromises synergistic actions of genotype-intrinsic differences and stress-induced responses.
2.2.4. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis of DEGs
Functional analysis of differentially expressed genes (DEGs) based on transcriptomic data revealed distinct gene regulation patterns between L. chinensis germplasm Lc71 (HR) and L. chinensis Lc5 (ES) germplasms following rust fungus infection. DEGs from the susceptible germplasm L. chinensis Lc5 (ES) were significantly enriched in biological processes, including starvation response (GO:0042594, 4.58%) and cell damage (GO:0001906, 2.37%). Upregulation of genes involved in amino sugar catabolism, such as hexosaminidase (Lc1Xm038572), could accelerate degradation of cell wall polysaccharides, potentially providing nutritional substrates like N-acetylglucosamine for the pathogen. At the molecular function level, genes associated with β-glucosidase (GO:0008422) and chitinase (GO:0004568) activities constituted 14.42% of DEGs (Figure 4a), and overactivation of cell wall hydrolases resulted in structural integrity loss. Meanwhile, the glycerolipid metabolism pathway exacerbated plasma membrane permeability alterations through membrane lipid hydrolysis, consistent with abnormal enrichment of plasma membrane-anchored component-related genes (GO:0046658) in cellular component analysis (Figure 4a).
DEGs in the L. chinensis Lc71 (HR) exhibited systematic defense regulation. In biological processes, genes involved in cell wall biogenesis (GO:0009832, 4.36%) and hemicellulose metabolism (GO:0010410, 3.2%) were significantly activated, and upregulation of cellulose synthase genes (e.g., Lc4Xm045201) promoted polysaccharide cross-linking network formation. Molecular function analysis revealed that genes associated with xyloglucan transferase (GO:0016762) and cellulose synthase activity (GO:0016759) constituted 3.06% of DEGs (Figure 4b), directly participating in physical barrier construction. Notably, although flavonoid biosynthesis was generally downregulated in HR, specific upregulation of isoflavone synthase genes (e.g., Lc2Xm054398) facilitated antimicrobial compound synthesis via branch-specific metabolic regulation.
Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis further revealed the molecular basis of resistance differentiation (Figure 5). While both germplasms showed significant enrichment in “metabolic pathways” and “biosynthesis of secondary metabolites,” expression patterns were antagonistic. In Lc71 (HR), DEGs were predominantly downregulated, potentially reducing metabolic consumption by suppressing secondary metabolism-related genes (e.g., flavonoids). Conversely, upregulated genes dominated in Lc5 (ES), where abnormal activation of monoterpenoid biosynthesis (ko00902) compromised metabolic homeostasis. This polarity indicates resource reallocation through competitive pathway suppression in HR, whereas metabolic disorder in ES exacerbated coactivation of defense genes and terpenoid synthesis. Germplasm-specific pathway analysis demonstrated that upregulated genes in Lc5 (ES)’s unique amino sugar/nucleotide sugar metabolism facilitated pathogen carbon acquisition, while Lc71 (HR) enriched pathways like plant circadian rhythm (ko04712) and isoflavonoid biosynthesis (ko00943) to coordinate defense gene timing and antimicrobial synthesis.
Cross-group comparisons revealed common enrichment of “plant-pathogen interaction” (ko04626) and “ABC (ATP-binding cassette) transporters” in 71RT vs. 5ST and 71RN vs. 5SN. Differential expression of resistance genes (e.g., TNL (Toll/interleukin-1 receptor-like nucleotide-binding leucine-rich repeat) family) in HR mediated pathogen recognition and hypersensitive response, whereas ABC transporter activation harbored roles in toxin efflux/defense molecule transport. Additionally, significant enrichment of suberin and cutin biosynthesis pathway in 71RT vs. 5ST, coupled with upregulated cell wall reinforcement genes, constituted a physical barrier defense layer. These results demonstrate that Lc71 (HR) achieves multi-level resistance through spatiotemporally transcriptional reprogramming—integrating cell wall fortification, defense compound synthesis, and pathogen recognition signaling. Conversely, metabolic pathway disorder in Lc5 (ES) exacerbated susceptibility.
2.3. Metabolomic Reprogramming Signatures in Rust-Infected L. chinensis Germplasms
2.3.1. Metabolomic Data and Quality Control (QC) of Samples
Quality control (QC) samples were prepared from pooled sample extracts. By analyzing the mass spectrometry results of different QC samples (total ion current, TIC, chromatograms), the reproducibility of metabolite extraction and detection can be assessed. As shown in Figure 6, TIC chromatograms exhibited high overlap with consistent retention times and peak intensities, indicating high signal stability across repeated mass spectrometer injections of identical samples.
2.3.2. Principal Component Analysis (PCA) of Metabolites and Validation of Orthogonal Partial Least Squares-Discriminant Analysis (OPLS-DA) Model
Principal component analysis (PCA) revealed distinct clustering among the 12 experimental samples. Quality control (QC) samples (n = 3) exhibited significant differences in metabolite composition (Figure 7). The first two principal components (PCs) cumulatively captured 56.55% of metabolic variation (PC1 = 40.83%, PC2 = 15.72%). L. chinensis Lc71 (HR) and L. chinensis Lc5 (ES) germplasms were completely separated along PC1 (Lc71 right, Lc5 left), indicating genotype-driven metabolite profile variation. Treatment vs. control groups were significantly differentiated along the PC2 axis (the treatment group was concentrated in the upper quadrant, while the control group was distributed in the lower quadrant), revealing that the metabolic reprogramming induced by rust fungus infection had commonalities between genotypes. The tight clustering of QC samples in the central area confirms the experimental reproducibility (relative standard deviation, RSD < 15%).
Orthogonal partial least squares-discriminant analysis (OPLS-DA) model validation showed (Table 2) demonstrated optimal explanatory power and predictive ability across all four comparison models: R^2^X = 0.602–0.729, indicating capture of 60.2–72.9% of metabolite variation; R^2^Y = 1.000, confirming completely resolution of inter-group differences; Q^2^ > 0.90 (range 0.913–0.952), affirming excellent predictive performance. These results met the validity threshold of Q^2^ > 0.50 in metabolomics studies and far exceeded the excellent standard of Q^2^ > 0.90, providing a reliable statistical basis for the subsequent screening of differentially accumulated metabolites (DAMs).
2.3.3. Screening of Differentially Accumulated Metabolites (DAMs)
Ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS) identified 2702 metabolites, revealing significant metabolic reprogramming in L. chinensis Lc71 (HR) and susceptible L. chinensis Lc5 following rust infection (Figure 8 and Figure 9). Intragroup comparisons showed the Lc5 (ES) treatment group (5ST vs. 5SN) contained 481 DAMs, with 393 (81.7%) significantly upregulated—primarily flavonoids (e.g., Lmgp005868), lipids (PD0677518), and lignans (Lamp007512)—while defensive substances such as phenolic acids (Zmhn002750) and coumarins (Zjgp122320) were downregulated (Figure 8a). In Lc71 (HR) treatment group (71RT vs. 71RN), 287 DAMs included 216 downregulated (75.3%) flavonoids (MWSHY0121) and amino acid derivatives (Lcsp001322), with upregulated alkaloids (Lcsp002898) and terpenoids (Zbzp003763) (Figure 8b), suggesting that it may save resources by suppressing basic metabolism and prioritize the synthesis of specific antimicrobial components.
Inter-group comparisons showed that in the non-inoculated control group (71RN vs. 5SN), there were 962 differential metabolites, with the Lc71 (HR) constitutively accumulating high levels of flavonoids (PD0209862 and others) and phenolic acids (Rlp03049); in the inoculated treatment group (71RT vs. 5ST), there were 865 differential metabolites, with the resistant germplasm specifically upregulating flavonoids and alkaloids, while downregulating competitive metabolites (Figure 8a). Venn analysis further revealed that Lc71 (HR) had 194 unique metabolites, including flavonoid derivatives such as 3,5-trimethoxyflavone glucoside, alkaloids such as caffeoyl-methyl-glucoside, and terpenoids such as apiol (Figure 9). These metabolites play key roles in plant defense, such as tricin 7-O-[feruloyl]-glucoside, which can inhibit the activity of rust fungal extracellular enzymes and hinder infection, and cis-p-coumaroyltyramine, which acts as a phytoalexin precursor involved in the hypersensitive response. In contrast, among the 388 unique metabolites of Lc5 (ES), fatty acid elongation products and monoterpenoids accounted for a significant proportion, and their abnormal accumulation may disrupt cell membrane homeostasis and interfere with normal defense metabolic flux.
The differences in metabolic profiles indicate that the HR optimizes defense by precisely regulating the secondary metabolic network: it suppresses the synthesis of broad-spectrum flavonoids to concentrate resources on the production of highly active antimicrobial compounds (such as nevadensin 7-rutinoside), while specifically accumulating cutin monomers (such as 1-O-feruloylquinic acid) to strengthen the physical barrier. In contrast, the metabolic disorder in ES leads to an imbalance in the synthesis and catabolism of defensive substances, ultimately creating a favorable environment for pathogen proliferation.
2.3.4. Kyoto Encyclopedia of Genes and Genomes (KEGG) Functional Annotation and Enrichment Analysis of Differentially Accumulated Metabolites (DAMs)
KEGG pathway enrichment analysis of DAMs revealed fundamentally distinct metabolic regulation strategies in resistant and susceptible L. chinensis germplasms post-rust infection (Figure 10). The ES (5ST vs. 5SN) upregulated the biosynthesis of luteolin aglycones, chrysoeriol aglycones, isoquinoline alkaloids, benzaldehyde derivatives, and glutathione metabolism, activating the synthesis of flavonoid antioxidants and enhancing glutathione peroxidase activity to alleviate oxidative damage (Figure 10a). The RH (71RT vs. 71RN) achieved precise defense through metabolic network reprogramming: it downregulated the pathways of stilbenoid, diarylheptanoid and gingerol biosynthesis, flavonoid biosynthesis, luteolin aglycones biosynthesis, and biosynthesis of p-coumaric acid derivatives, inhibiting the synthesis of broad-spectrum antimicrobial substances; meanwhile, it upregulated the pathways of cutin, suberine and wax biosynthesis, nucleotide metabolism, and pyrimidine metabolism (Figure 10b).
In the comparison group of 71RT vs. 5ST (Figure 10c), DAMs were enriched and upregulated in the pathways of luteolin aglycones biosynthesis and biosynthesis of phenylacetic acid derivatives, while they were enriched and downregulated in the pathways of biosynthesis of protocatechuic acid derivatives, stilbenoid, diarylheptanoid and gingerol biosynthesis, chrysoeriol aglycones biosynthesis, flavonoid biosynthesis, biosynthesis of flavones aglycones III, biosynthesis of caffeic acid derivatives, biosynthesis of p-coumaric acid derivatives, and biosynthesis of ferulic acid derivatives.
Overall, the susceptible germplasm attempted to resist the pathogen by upregulating flavonoid compounds and glutathione metabolism, activating basic defense metabolism and the antioxidant system. In contrast, the resistant germplasm downregulated the pathways of stilbenoids; flavonoids; p-coumaric/caffeic acid derivatives; and biosynthesis of pantothenate and CoA, while upregulating the pathways of cutin, suberine, and wax biosynthesis, and nucleotide and pyrimidine metabolism. It is speculated that it resists the pathogen through resource optimization strategies, strengthening physical barriers, and inhibiting pathogen nutrient acquisition.
2.4. Coordinated Transcriptomic Metabolomic Dynamics During Rust Infection
2.4.1. KEGG Pathway Co-Enrichment Analysis
Co-enrichment analysis of differentially expressed genes (DEGs) and DAMs revealed interactive networks in response to rust infection (Figure 11). In the 71RT vs. 71RN comparison group, DEGs and DAMs were co-enriched in 39 KEGG pathways, among which the luteolin biosynthesis and flavonoid biosynthesis metabolic pathways showed relatively higher enrichment of both DAMs and DEGs compared to other pathways. In the flavonoid biosynthesis pathway, DEGs were extremely significantly enriched (p < 0.01), while DAMs were enriched but not significantly (p > 0.05).
The Lc71 (HR) had unique enriched pathways including flavonoid and flavonol biosynthesis, isoflavonoid biosynthesis, betaine biosynthesis, tyrosine biosynthesis, isoquinoline alkaloid biosynthesis, chrysoeriol aglycones biosynthesis, and cofactor biosynthesis pathways. The Lc5 (ES) had unique enriched pathways in phosphatidylinositol signaling system, lysine degradation, zeatin biosynthesis, linoleic acid metabolism, and glutathione metabolism.
To further elucidate the resistance mechanism, this study selected the flavonoid biosynthesis pathway, which was enriched in both DAMs and DEGs after inoculation in both resistant and susceptible L. chinensis, and the nucleotide metabolism pathway, which showed upregulation in DAMs, for in-depth analysis.
2.4.2. Expression Correlation Analysis
Canonical correlation analysis (CCA) was employed to elucidate the regulatory network of DEGs and DAMs in the flavonoid biosynthesis (ko00941) and nucleotide metabolism (ko01232) pathways in Lc71 (HR) at 48 h post-inoculation with rust fungus (Figure 11). In the flavonoid pathway (ko00941), genes Lc2Xm054395 and Lc6Xm080166 were significantly positively correlated with phenolic acid compounds chlorogenic acid and 5-O-caffeoylquinic acid, with their expression levels being downregulated in concert. Meanwhile, genes Lc2Xm054924, Lc4Xm044161, novel.8850, Lc2Ns006303, and Lc7Ns021884 exhibited significant positive correlations with flavonoids naringenin and naringenin 7-O-beta-D-glucoside (Figure 12a), all showing a downward trend. Notably, gene Lc7Xm040717 showed a negative correlation with the aforementioned metabolites.
In the nucleotide metabolism pathway (ko01232), genes Lc5Ns011910, Lc1Xm057211, Lc4Xm043884, mws0255, and pme1194 showed negative correlations with the corresponding metabolites, with gene downregulation accompanied by metabolite accumulation. In contrast, genes Lc1Xm057209 and pmb0981 exhibited negative correlations, characterized by gene upregulation and metabolite downregulation (Figure 12b). These genes play important roles in the flavonoid biosynthesis and nucleotide metabolism pathways, and Lc71 (HR) may respond to pathogen infection by regulating these genes and metabolites in the pathways.
3. Discussion
3.1. Evaluation of Rust Resistance in Leymus chinensis
Leymus chinensis, a native grass species in China, serves not only as an excellent forage but also exhibits multiple stress-resistant traits, including cold resistance, drought tolerance, and saline–alkaline adaptation [42]. As a major disease affecting L. chinensis, rust prevalence severely compromises its production and grassland ecological balance [43]. Consequently, evaluating rust resistance in L. chinensis germplasm resources and exploring associated resistance genes are critical for improving Poaceae crops and forage grasses [44]. Nevertheless, systematic assessment of rust resistance in Chinese L. chinensis germplasm resources remains unavailable. Utilizing 24 germplasms previously collected by our team, this study conducted indoor rust resistance evaluations. The incidence rate (6.67–100%) and disease index (DI) (1.33–80.87) revealed significant resistance variation among germplasms from different geographical origins, consistent with findings by Zhang [9], Miedaner [45], and Liu [46]. Six highly resistant (HR) germplasms were identified: Lc30, Lc53, Lc62, Lc68, Lc70, and Lc71. We posit that their resistance is predominantly attributable to genetic factors, suggesting these accessions may harbor broad-spectrum resistance genes and serve as core breeding materials.
3.2. Transcriptomic Analysis of Resistant and Susceptible L. chinensis in Response to Rust Infection
Zeng et al. [47] analyzed transcriptomes of a Tibetan hulless barley germplasm with high powdery mildew resistance. Compared to controls, differentially expressed genes (DEGs) in resistant germplasm were enriched in cell wall synthesis, secondary metabolite biosynthesis, signal transduction, and photosynthesis. Similarly, Bilgin et al. [48] observed frequent downregulation of photosynthesis-related genes across eight plant species under 22 biotic stresses, suggesting an adaptive defense strategy via resource reallocation. Saha et al. [49] mapped rust resistance genes in lentil germplasm using molecular markers. They observed that upon inoculation, resistant germplasm rapidly upregulated cell wall-associated genes (e.g., peroxidase, PRX; peroxidase precursor, PER) and photosynthesis-related genes, whereas susceptible germplasm exhibited enhanced expression of fatty acid metabolism genes. Yan et al. [50] applied mesophyll single-cell analysis to rust-resistant and susceptible maize inbred germplasms. Their results demonstrated that at 48 hours post-inoculation (hpi), susceptible germplasm primarily reprogrammed fatty acid metabolism and monoterpene biosynthesis to combat rust stress, while resistant germplasm maintained homeostasis through cell wall thickening and photosynthetic regulation.
Consistent with these findings, our study revealed that following rust fungus infection, Lc5 (extremely susceptible, ES) responded primarily by regulating cellular components and primary metabolic processes, such as fatty acid metabolism and monoterpene biosynthesis. Lc71 (HR), however, regulated the biosynthesis of metabolites, cell wall synthesis, and photosynthesis-related processes to sustain essential physiological functions and combat rust fungus stress. These results indicate that distinct germplasms employ different strategies against rust infection, with the resistant germplasm placing greater emphasis on sustaining overall plant health status and defense capabilities.
3.3. Metabolomic Analysis of Resistant and Susceptible L. chinensis in Response to Rust Infection
Metabolomic analysis revealed significant divergence in metabolic regulation between rust-resistant germplasm Lc71 (HR) and Lc5 (ES) following rust fungus infection. Flavonoid compounds (flavones, isoflavones, etc.) typically function in stress responses by (1) scavenging reactive oxygen species (ROS) and neutralizing free radicals [51]; (2) synthesizing antimicrobial phytoalexins such as luteolin [52]; or (3) interacting with transcription factors to activate immune genes [53,54]. Notably, Huang et al. [55] demonstrated that moderate flavonoid reduction activates alternative defense mechanisms, enhancing Gossypium hirsutum resistance to Fusarium wilt. Similarly, Zuo Tao et al. [56] suppressed catechin synthesis via antisense dihydroflavonol reductase (DFR) expression in poplar, confirming that controlled flavonoid depletion can improve resistance against specific pathogens. Nucleotide metabolism pathways play diverse roles in plant disease resistance, as they can act as signaling molecules directly involved in disease resistance, provide energy and synthetic precursors to support resistance responses, regulate gene expression, and interact with other metabolic pathways such as plant hormone metabolism to collectively enhance plant resistance [57,58,59]. In this study, the Lc5 (ES) attempted to resist the pathogen by upregulating flavonoid compounds and glutathione metabolism, activating the basic defense metabolism and antioxidant system to prevent pathogen infection. Lc71 (HR), on the other hand, downregulated the pathways of stilbenoids, flavonoids, p-coumaric/caffeic acid derivatives, and biosynthesis of pantothenate and CoA, while upregulating nucleotide and pyrimidine metabolism, cutin, suberine, and wax biosynthesis. These findings align with conserved defense signatures reported by Mashabela et al. [60] in Poaceae. Specifically, Lc71 demonstrated rapid mobilization of phenylpropanoid-flavonoid metabolism (e.g., luteolin-7-O-glucuronide accumulation), whereas susceptible Lc5 exhibited dominant fatty acid/terpenoid synthesis. This metabolic bifurcation suggests that preferential phenylpropanoid activation may represent a cross-species resistance indicator in monocot-Puccinia pathosystems, warranting validation across diverse host–pathogen combinations.
3.4. Integrated Transcriptomic and Metabolomic Analysis of Resistant and Susceptible L. chinensis in Response to Rust Infection
Zhang et al. [58] reported that during late-stage rust infection in rice 48–120 hpi, activation of the jasmonic acid/systemic acquired resistance (JA/SAR) pathway triggered significant enrichment in the phenylpropanoid-lignin biosynthesis pathway, nucleotide sugar metabolism, and tryptophan metabolism. This response led to host cell wall thickening and the formation of continuous lignified rings. However, no significant alterations in fatty acid or monoterpenoid metabolism were detected. Integrated multi-omics analysis demonstrated that the resistant germplasm Lc71 (HR) establishes a multi-tiered defense network via gene–metabolite coregulation. Within the flavonoid biosynthesis pathway (ko00941), the hydroxylase gene Lc2Xm054395 and the 4-coumarate-CoA ligase gene Lc6Xm080166 exhibited significant positive correlations with chlorogenic acid and 5-O-caffeoylquinic acid, both being downregulated. This coordinated suppression likely reduces phenolic acid accumulation through inhibition of phenylpropane metabolism, consequently enhancing cell wall lignification. Simultaneously, glycosyltransferase gene Lc7Xm040717 showed a negative correlation with naringenin glycosides; its upregulation may redirect metabolic precursors toward alternative defense branches via glycosylation modifications.
Cross-group comparisons (71RT vs. 5ST) revealed 79 co-enriched pathways. In the nucleotide metabolism pathway (ko01232), genes Lc5Ns011910, Lc1Xm057211, Lc4Xm043884, and UTP analog mws0255 displayed negative correlations with metabolite pme1194, characterized by concurrent gene downregulation and metabolite accumulation. Conversely, genes Lc1Xm057209 and pmb0981 demonstrated inverse regulation patterns with gene upregulation coupled to metabolite depletion. These interactions suggest Lc71 (HR) modulates pathogen response through precise pathway regulation.
Beyond these core pathways, differentially expressed genes (DEGs) in Lc71 (HR) were significantly enriched in flavonoid/flavonol biosynthesis, isoflavonoid biosynthesis, betaine biosynthesis, tyrosine biosynthesis, and isoquinoline alkaloid biosynthesis. Corresponding differentially accumulated metabolites (DAMs) preferentially accumulated in chrysoeriol aglycone biosynthesis and cofactor biosynthesis pathways. In contrast, susceptible Lc5 (ES)-specific DEGs enriched phosphatidylinositol signaling, lysine degradation, zeatin biosynthesis, linoleic acid metabolism, and amino sugar metabolism pathways, with its DAMs dominating glutathione metabolism. Critical genes and metabolites within these pathways constitute pivotal components of disease resistance mechanisms, offering molecular targets for rust-resistant L. chinensis breeding.
4. Materials and Methods
4.1. Materials
The tested rust fungus Ycgx-2 was isolated from rust-infected Leymus chinensis leaves collected at the experimental base in Youyu County, Shanxi Province. This pathogen was provided and preserved by the College of Grassland Science, Shanxi Agricultural University.
The 24 assessed L. chinensis germplasms (Table 3), originating from heterogeneous habitats across northern China and Mongolia, underwent standardized cultivation using a polyvinyl chloride (PVC) pipe-based system. Germplasms with differential rust resistance were selected based on prior field phenotypic evaluations. In 2022, tiller propagation from identical mother plants facilitated the transplantation of these germplasms into a climate-controlled greenhouse. The specific methodologies are as follows:
A sterilized substrate mixture of garden soil and vermiculite (1:1, v/v) was prepared through high-pressure moist heat sterilization (121 °C for 25 min, repeated twice). After cooling, the mixture was sealed and stored under sterile conditions. Healthy plants underwent standardized processing: aerial parts were excised 3 cm aboveground using sterile scissors, while rhizomes were stripped and segmented into 5–10 cm sections containing ≥2 stem nodes. Cultivation employed 15 × 15 × 15 cm pots following this protocol: a sterile moist towel was layered at the pot base; sterilized substrate filled two-thirds of the volume; rhizome segments were inserted at original cultivation depth with repeated pot-lifting to compact soil and ensure root–soil contact; after planting, Hoagland nutrient solution was applied thoroughly. Post-acclimation, substrate moisture was maintained at 75 ± 5%. Plants were grown in climate chambers under 16/8 h (light/dark) photoperiod at 25 ± 3 °C, with 50 mL Hoagland solution (Qingdao Haibo Biotechnology Co., Ltd., Qingdao, China; Cat: HB8870) was applied thoroughly. After one year, transplantation to 10×10×8 cm square pots were implemented using a completely randomized block design with three biological replicates per germplasm (6–9 plants per pot).
4.2. Evaluation of Rust Resistance in 24 Leymus chinensis Germplasms Under Laboratory Conditions
Ten days prior to inoculation, aerial parts of L. chinensis were excised 3 cm above ground. Upon reaching the one-leaf-one-heart stage in regenerated foliage, a rust spore suspension (1 × 10^5^ spores·mL^−1^) was prepared following Reference [61]. Using a handheld pressure sprayer, all 24 germplasms were inoculated until uniform mist films formed on leaf surfaces without droplet formation.
Post-inoculation dew chamber conditions implemented a gradient humidity protocol: 20 min atomization/60 min interval (2 cycles), 5 min atomization/60 min interval (4 cycles), and 1 min atomization/120 min interval, maintaining ≥95% relative humidity for 24 h. Plants were then transferred to sterile culture rooms at 25–28 °C, 45–50% RH, and 16/8 h (light/dark) photoperiod. Hoagland nutrient solution (20 mL/pot) was applied on days 3 and 10 post-inoculation, with regular tiller removal. Disease progression was monitored daily, with severity assessed visually on days 7 and 12 based on maximum symptom expression.
Disease severity grading followed a six-level standard adapted from (NY/T 1443–2007) [62], incorporating L. chinensis-specific rust characteristics: Level 0 (no disease), Level 1 (0–5% leaf area covered by urediniospores), Level 2 (5–25%), Level 3 (25–50%), Level 4 (50–75%), and Level 5 (75–100%).
The formula for calculating the diseased leaf rate is as follows:
The formula for calculating the disease index (DI) is as follows:
In Equation (2): DI is the disease index; i is the disease grade; X_i_ is the number of units at grade i; S_i_ is the representative value of severity at grade i; S_max_ is the maximum severity value.
The six-tiered classification standard for L. chinensis rust resistance (Table 4) was established by integrating the disease resistance grading methodologies of Festuca arundinacea [63], Bletilla striata [64], and Poa pratensis [65], with two-year field dynamic monitoring data. The mean DI across both years served as the primary grading benchmark. Genotypes classified as ‘ES’ were determined based on severe symptom expression and high pathogen load, consistent with standard plant pathology practices [66].
4.3. Inoculation of Rust Fungi and Sampling in Resistant and Susceptible L. chinensis
The contrasting germplasms Lc71 (HR) and Lc5 (ES) were selected for subsequent analysis. After 20 days of pre-cultivation at 3.00 cm stubble height, seedlings were inoculated at the seedling stage following Section 4.2 protocols: treatment groups received Ycgx-2 urediniospore suspension (0.05% Tween-20), while controls received sterile water (0.05% Tween-20), with three biological replicates per treatment. At 24 hours post-inoculation (hpi), plants were transferred to sterile culture rooms. Leaf samples collected at 48 hpi were pooled equally from three replicates and divided for transcriptome sequencing (0.30 g) and wide-targeted metabolomics (0.60 g) [46,67]. Samples were labeled as 5ST (Lc5 treated), 71RT (Lc71 treated), 5SN (Lc5 control), and 71RN (Lc71 control), flash-frozen in liquid nitrogen, and stored at −80 °C. Successful inoculation was confirmed by typical urediniospore pustules on Lc5 leaves at 7 days post-inoculation (dpi) and significant symptom expansion at 10 dpi (controls remained asymptomatic), after which samples were submitted to Wuhan Metware Biotechnology Co., Ltd. (Wuhan, China) for omics analysis.
4.4. Transcriptome Sequencing and Data Analysis
Total RNA was extracted from L. chinensis leaves using the cetyltrimethylammonium bromide-PBIOZOL (CTAB-PBIOZOL) combined method (Biozol Reagent, BioFlux Co., Ltd., Hangzhou, China; RNA integrity number, RIN ≥ 7.0), with concentration quantified by Qubit 4.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and integrity verified via Qsep400 Bio-Fragment Analyzer. (Bioptic Inc., New Taipei, Taiwan, China). Strand-specific complementary DNA (cDNA) libraries were constructed using the Illumina TruSeq™ Stranded mRNA Kit (Illumina Inc., San Diego, CA, USA): poly(A) + mRNA was enriched by Oligo(dT) beads and fragmented in Mg^2+^ buffer (94 °C, 200–300 bp); double-stranded cDNA (ds-cDNA) was synthesized with SuperScript™ IV Reverse Transcriptase using deoxyuridine triphosphate (dUTP) for strand marking; TruSeq™ adapters were ligated and 250–350 bp inserts selected via 2% agarose gel electrophoresis; final amplification involved 15 polymerase chain reaction (PCR) cycles with Phusion™ High-Fidelity DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA).. Qualified libraries (size peak: 280 ± 14 bp, coefficient of variation, CV < 5%) underwent paired-end 150 bp (PE150) sequencing on Illumina NovaSeq™ 6000 (Illumina Inc., San Diego, CA, USA; Q30 ≥ 85%).
Bioinformatics analysis followed Wuhan Metware Biotechnology Co., Ltd.’s standard RNA-seq pipeline: (1) Raw data filtering by fastp v0.23.2 (remove adapters, Q20 < 50%, N > 10%, retain Q30 ≥ 85% reads). (2) Alignment to the L. chinensis reference genome [68] (dataset doi:10.6084/m9.figshare.24032238) via HISAT2 v2.2.1 (parameters: --n_base_limit 15 --qualified_quality_phred 20; with alignment rate ≥ 85%). (3) Transcript assembly with StringTie v2.2.6 and novel transcript prediction by Coding Potential Calculator 2 (CPC2) v0.1. (4) Gene expression quantification using featureCounts v2.0.3 (default parameters; FPKM normalization) and differentially expressed genes (DEGs) screening via DESeq2 v1.22.1 (absolute log_2_ fold-change |log_2_FC| ≥ 1, Benjamini–Hochberg false discovery rate FDR < 0.05). (5) Gene Ontology/Kyoto Encyclopedia of Genes and Genomes (GO/KEGG) enrichment with clusterProfiler v4.6.0 (hypergeometric test, FDR < 0.05).
4.5. Metabolome Sequencing and Data Analysis
Leaf samples were flash-frozen in liquid nitrogen, lyophilized at −50 °C for 63 h, and homogenized to 200 μm powder using a ball mill (Retsch GmbH, Haan, Germany). Exactly 50 mg powder was extracted with 1.2 mL of pre-chilled 70% methanol containing 250 μg/mL lidocaine (internal standard; Sigma-Aldrich, St. Louis, MO, USA). After vortexing and centrifugation, supernatants were filtered through 0.22 μm membranes (Merck Millipore, Burlington, MA, USA) for ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS) analysis. Chromatography employed an Agilent SB-C18 column (2.1 × 100 mm, 1.8 μm; Agilent Technologies, Santa Clara, CA, USA) with mobile phases: A = 0.1% formic acid/water, B = 0.1% formic acid/acetonitrile. The gradient program ran: 5% B (0 min) → 95% B (9 min) → 95% B (10 min) → 5% B (11.1 min) → 5% B (14 min) at 0.35 mL/min, 40 °C. Mass spectrometry used electrospray ionization ± mode switching (ESI ±) (ion spray voltage: ±5500 V/−4500 V; source temperature: 500 °C; curtain gas: 25 psi; nebulizer gas: 50 psi) with multiple reaction monitoring (MRM) transitions monitored on quadrupole-linear ion trap mass spectrometry (QTRAP-MS). Data processing involved: peak area extraction by SCIEX OS 3.0 software (AB Sciex, Framingham, MA, USA); Total Useful Signal normalization; Differential metabolite (DM) screening via orthogonal partial least squares-discriminant analysis (OPLS-DA) (R^2^Y > 0.90, Q^2^ > 0.50; thresholds: VIP > 1.0, |log_2_FC| ≥ 1.0, FDR < 0.05), and pathway analysis through KEGG annotation with hypergeometric test (p < 0.05) and impact score filtering (> 0.10).
5. Conclusions
This study systematically deciphers the molecular basis of rust resistance in Leymus chinensis through integrated transcriptomic and metabolomic profiling of contrasting germplasms. Evaluation of 24 geographically diverse accessions identified six highly resistant (HR) (e.g., Lc71) and five extremely susceptible (ES) (e.g., Lc5) genotypes, revealing that HR germplasms orchestrate resistance by prioritizing resource allocation to structural defense and nucleotide signaling. Key mechanisms include the following: (1) Upregulation of cell wall biosynthesis genes (Lc2Xm054395, Lc6Xm080166) with concurrent suppression of flavonoid pathways in HR germplasms (1012 differentially expressed genes (DEGs)); (2) Metabolic shift toward cutin synthesis (e.g., 1-O-feruloylquinic acid) reinforcing physical barriers; and (3) Gene–metabolite coregulation in 79 enriched pathways, notably Lc2Xm054395/chlorogenic acid negative correlation enhancing lignification and nucleotide metabolism fueling defense. These findings provide molecular targets for breeding. Future work should validate candidates (e.g., Lc7Xm040717) via CRISPR-Cas9, dissect cutin-phenylpropanoid crosstalk in field environments, and explore nucleotide signaling in monocot immunity.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Li Y.X. Ma H.Y. Ni H.W. Li S.Y. Xu L. Sun M.D. Qi W.W. Zhao D.D. Adaptation Responses of Different Ecotypes of Leymus chinensis to Saline–Alkaline Stress Front. Ecol. Evol.202412136112410.3389/fevo.2024.1361124 · doi ↗
- 2Wu Z.N. Hou X.Y. Ren W.B. Wang Z.L. Chang C. Yang Y.P. Yang Y.T. Prediction of the Potential Geographic Distribution of Leymus chinensis Based on Max Ent and Collection and Protection of Germplasm Acta Pratacult. Sin.201827125135
- 3Guo H.H. Hao M.D. Meng J. Xiao Q.H. Wu D.B. Liu G.S. Fertilization Effects on Grass Yield and Nutrient Uptake of Leymus chinensis in Artificial Grasslands J. Ningxia Univ. Nat. Sci. Ed.201637118124
- 4Niu Y. Yang S. Wang G. Liu L. Hua L. Effects of Grazing Disturbance on Plant Diversity, Community Structure and Direction of Succession in an Alpine Meadow on Tibet Plateau, China Acta Ecol. Sin.20183817918510.1016/j.chnaes.2018.02.007 · doi ↗
- 5Li B. Dewey C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome BMC Bioinform.20111232310.1186/1471-2105-12-32321816040 PMC 3163565 · doi ↗ · pubmed ↗
- 6Zhang X. You Y. Wang D. Zhu L. Quality Evaluation of the Soil-Root Composites Layer of Leymus chinensis Grassland Based on Different Degradation Degrees Catena 202221510633010.1016/j.catena.2022.106330 · doi ↗
- 7Hovmøller M.S. Sørensen C.K. Walter S. Justesen A.F. Diversity of Puccinia striiformis on Cereals and Grasses Annu. Rev. Phytopathol.20114919721710.1146/annurev-phyto-072910-09523021599494 · doi ↗ · pubmed ↗
- 8Gultyaeva E.I. Bespalova L.A. Ablova I.B. Shaydayuk E.L. Khudokormova Z.N. Yakovleva D.R. Titova Y.A. Wild Grasses as the Reservoirs of Infection of Rust Species for Winter Soft Wheat in the Northern Caucasus Vavilovskii Zhurnal Genet. Selektsii 20212563864610.18699/VJ 21.07234782883 PMC 8558919 · doi ↗ · pubmed ↗