De Novo Hybrid Assembly of the Tripterygium wilfordii Mitochondrial Genome Provides the Chromosomal Mitochondrial DNA Structure and RNA Editing Events
Yisha Cai, Suxin Yang, Haimei Chen, Yang Ni, Jingling Li, Jinghong Zhang, Chang Liu

TL;DR
This study assembles the mitochondrial genome of Tripterygium wilfordii and identifies its structure and RNA editing events, offering insights into its evolution and potential for genetic breeding.
Contribution
The first de novo hybrid assembly of the T. wilfordii mitochondrial genome, revealing its chromosomal structure and RNA editing sites.
Findings
The T. wilfordii mitogenome is 720,306 bp and encodes 55 genes, including 35 protein-coding genes.
Phylogenetic analysis shows T. wilfordii is closely related to Euonymus alatus.
600 RNA-editing sites were identified, with ccmB and nad4 genes having the highest number.
Abstract
Tripterygium wilfordii has extremely important pharmaceutical value in both traditional and modern medicine. The mitogenome of T. wilfordii was subjected to assembly and annotation with Nanopore long reads and Illumina short reads in this study. The mitogenome is 720,306 bp in length and is responsible for encoding 55 specific genes, including 35 protein-coding genes (PCGs), 17 transfer RNA (tRNA) genes, and 3 ribosomal RNA (rRNA) genes. Upon repetitive sequence analysis, 223 simple sequence repeats (SSRs), 24 long tandem repeats (LTRs), and 47 dispersed repetitive sequences (DRSs) were identified. The 24 common PCGs were used for phylogenetic analysis, which revealed that T. wilfordii is more closely related to Euonymus alatus. Moreover, mitochondrial plastid DNA (MTPT) analysis revealed eight MTPTs in the mitochondrial genome. Furthermore, 600 RNA-editing sites were detected in the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
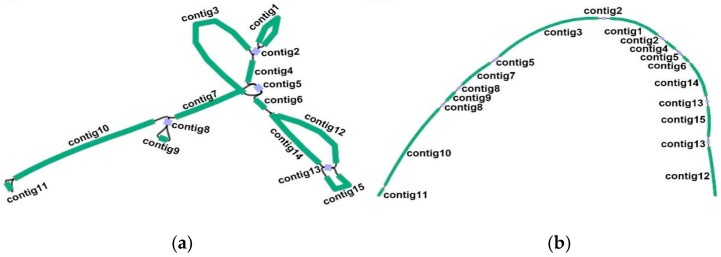 Figure 1
Figure 1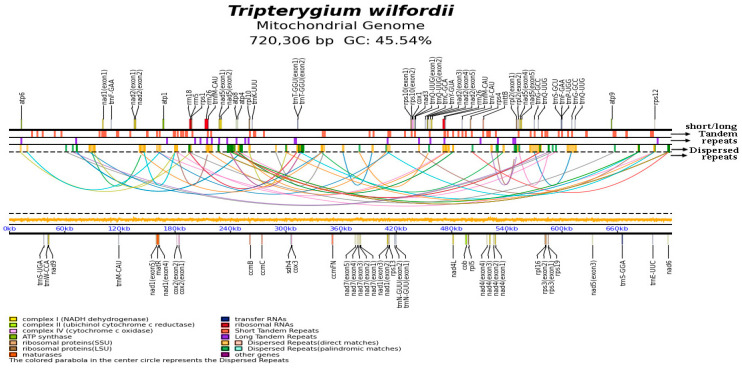 Figure 2
Figure 2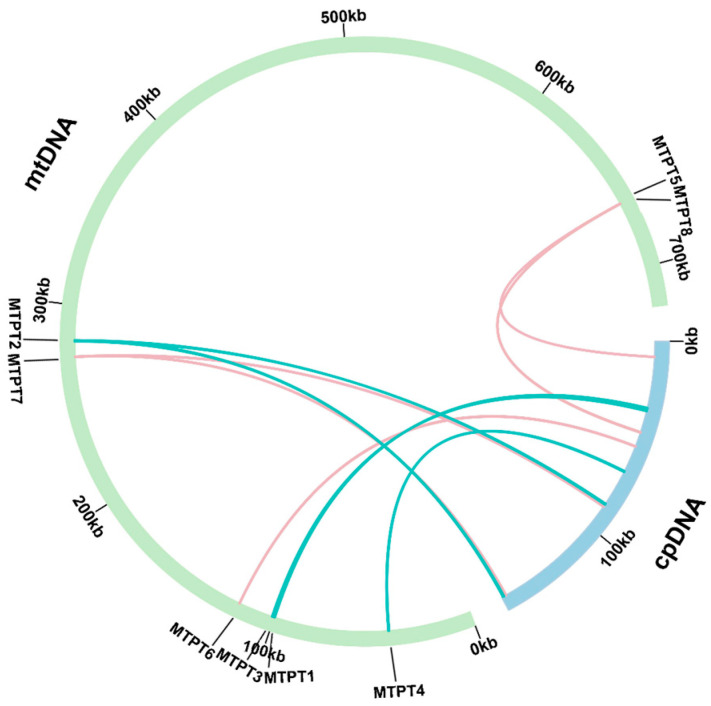 Figure 3
Figure 3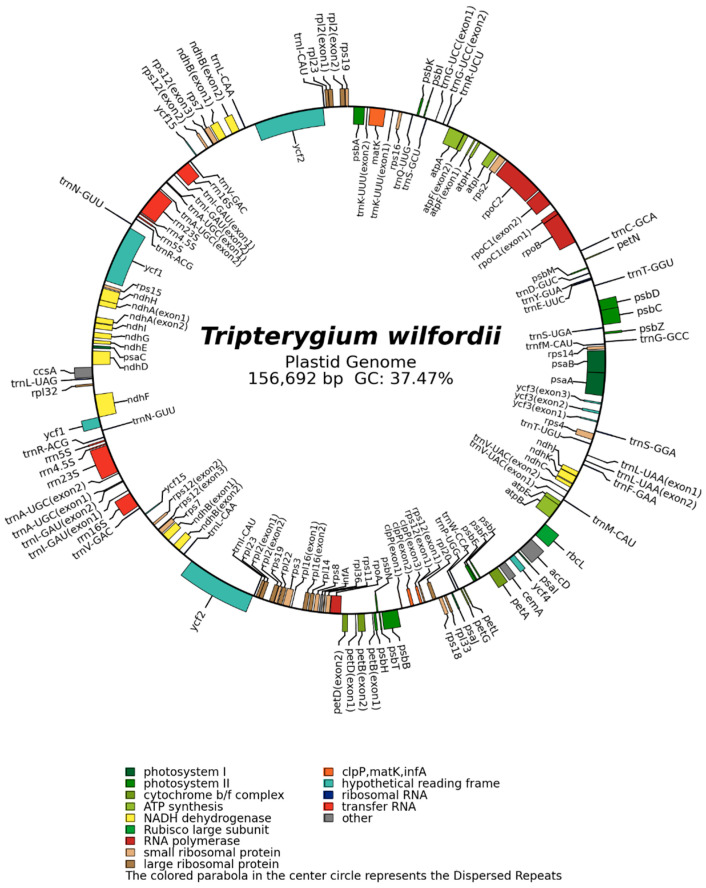 Figure 4
Figure 4 Figure 5
Figure 5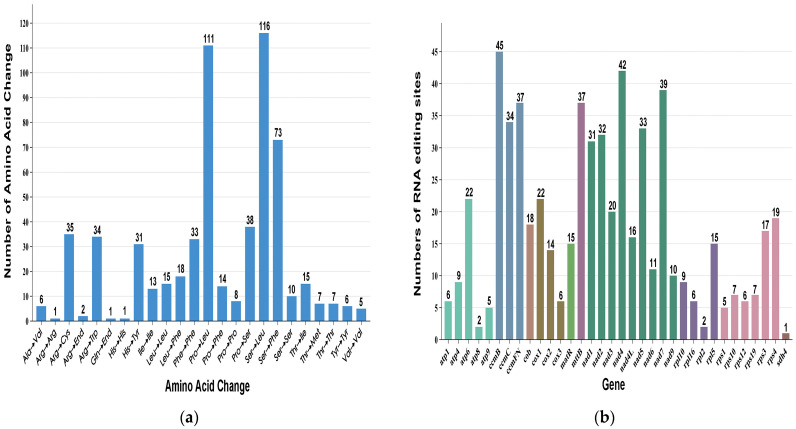 Figure 6
Figure 6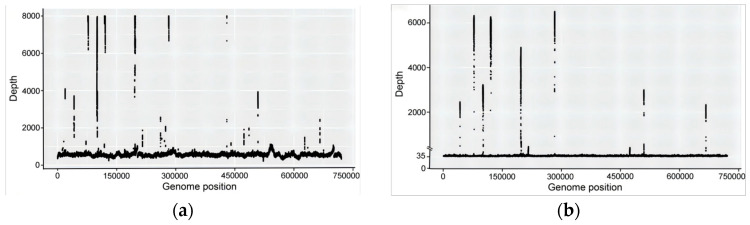 Figure 7
Figure 7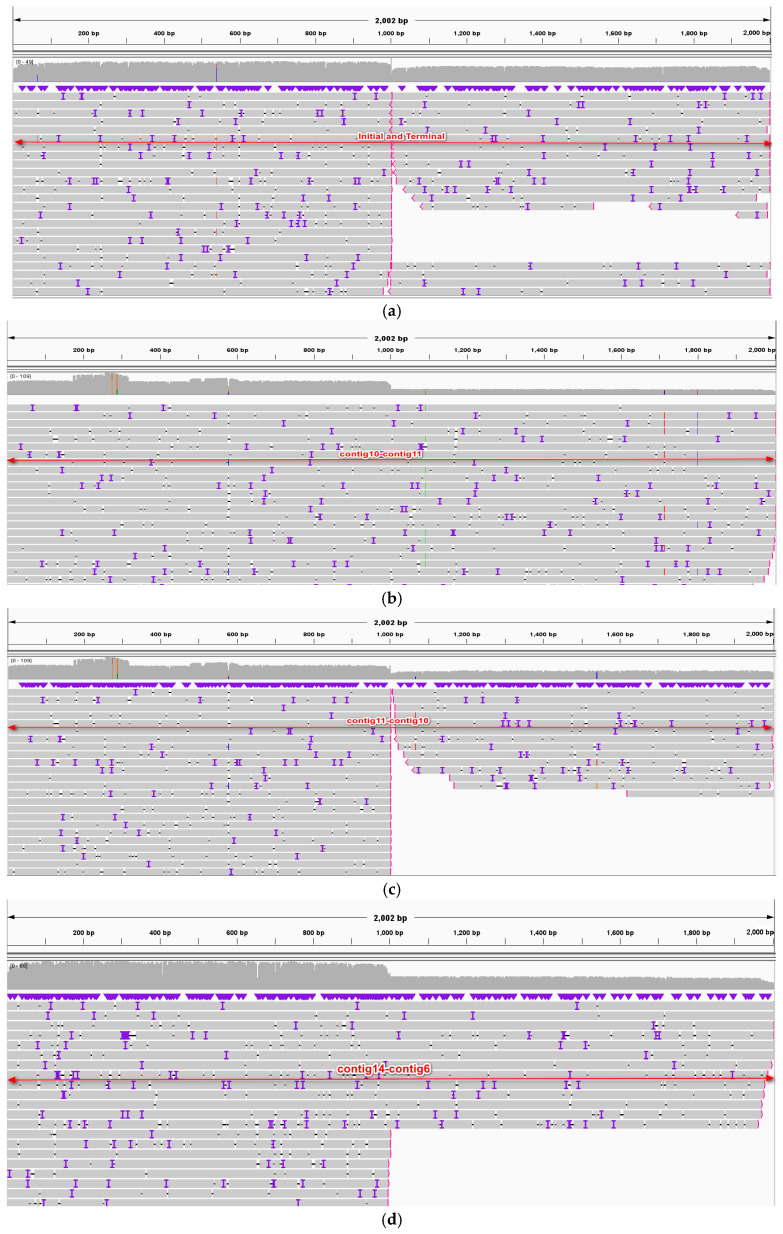 Figure 8
Figure 8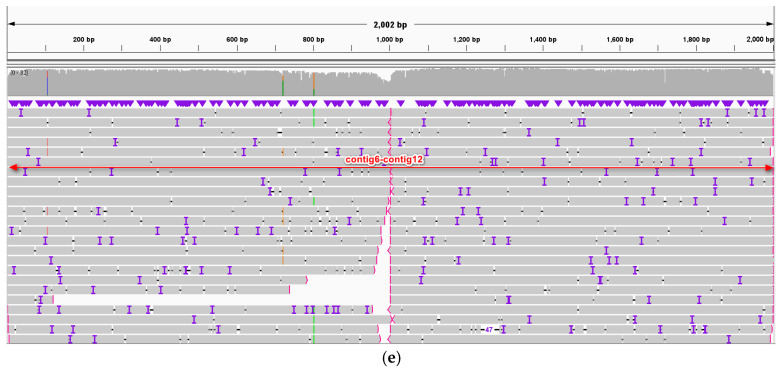 Figure 9
Figure 9- —National Science & Technology Fundamental Resources Investigation Program of China
- —Guiding projects in Fujian Province
- —Natural Science Foundation of Xiamen, China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNatural Compounds in Disease Treatment · Chromosomal and Genetic Variations · Natural product bioactivities and synthesis
1. Introduction
Tripterygium wilfordii Hook. f. is a perennial twining shrub belonging to the Celastraceae family and is distributed widely across East Asia [1]. The species is recognized as a Chinese medicinal herb that is used for managing various autoimmune conditions, such as rheumatoid arthritis and systemic lupus erythematosus [2,3]. T. wilfordii has numerous secondary metabolites, including triptolide and celastrol, which possess anti-inflammatory, immuno-suppressive, and antitumor activities and, therefore, have great development potential in the field of modern medicine [4,5]. Moreover, triptolide and celastrol are important in managing central nervous system disorders, such as Parkinson’s disease and Alzheimer’s disease [6,7]. Celastrol can also be used for the treatment of metabolic disturbances, such as high-fat-diet-induced obesity and type 2 diabetes [8,9]. High-throughput sequencing substantially contributes to the study of plant transcriptomes and genome architecture and is also useful when exploring plant genetic variations and secondary metabolites at the molecular level. Many transcriptome and genome studies conducted to date have revealed the key enzyme-encoding genes in the mevalonate (MVA) and methylerythritol phosphate (MEP) pathways, which regulate the biosynthesis of triptolide and celastrol through transcriptome data, as well as various transcription factors or miRNAs that regulate the biosynthesis of secondary metabolites in T. wilfordii, such as the negative regulation of triptolide biosynthesis by MYC2 [10]. In addition, the high-quality reference T. wilfordii genome has been illustrated, highlighting the regulatory role of CYP450 in the biosynthesis of triptolide [11]. The elucidation of the complete T. wilfordii chloroplast genome will be useful for molecular-assisted breeding in future studies [1]. However, the T. wilfordii mitochondrial genome sequence remains unreported to date, greatly limiting subsequent studies on this species.
In plants, the mitochondrial genome, which has many kinds of rearrangements, can be quite complex, although it may be used for various beneficial applications. First, as the mitochondrial genome has an extremely high rate of evolution, it has been extensively used as a molecular marker in modern molecular biology research, such as for reconstructing the phylogenetic relationships of species. Second, as cellular organelles, mitochondria are responsible for generating ATP and various metabolites, and the mitochondrial release of the metabolites of the tricarboxylic acid (TCA) cycle controls cell activity and fate [12]. Third, the plant mitogenome is huge and has a complicated structure and genetic diversity, although the stability of a plant mitogenome is protected through certain DNA repair mechanisms, particularly homologous recombination (HR) and base excision repair (BER) [13]. Finally, RNA editing, such as the conversion of cytidines (C) to uridines (U), which is important for the normal functioning of protein-encoding genes, is frequently detected in plant mitochondria [14]. In addition, T. wilfordii has long been used for asexual reproduction, resulting in many problems, such as confusion in terms of variety, serious degradation, and a low content of active ingredients. Consequently, exploring the T. wilfordii mitochondrial genome is necessary. This work, therefore, involved the sequencing and assembly of the complete T. wilfordii mitochondrial genome; analysis of the potential T. wilfordii mtDNA structure; and comparative analyses of repetitive sequences, homologous recombination, phylogenetic relationships, and RNA editing for this species. The findings of this study are expected to contribute to the further exploration of scientific issues encountered in relevant research at the molecular level and lay a certain theoretical basis for the genetic engineering and breeding of medicinal plants.
2. Results
2.1. Graph-Based Mitogenome and General Genomic Features
The hybrid assembly method was used to construct the T. wilfordii mitogenome by adopting long/short reads. First, 10 Gb of Illumina short reads and 7 Gb of Nanopore long reads of the total DNA were sequenced (Table A1). GetOrganelle software (v1.7.0) was then used for extracting and elongating the mitochondrial genome short reads, and SPAdes software (v3.8.2) in Unicycle was usyed for assembly into a unitig graph using the whole-genome sequencing results (Figure 1a). Moreover, Illumina short reads together with Nanopore long reads were used for calculating the sequencing depth at each genomic locus, thereby validating the accuracy of the assembly. The sequencing depth remained consistent among the genomic locations, without detectable gaps (Figure A1a,b). The T. wilfordii mitochondrial genome was revealed to have 15 contigs, including 4 double-bifurcation structures (Table 1 and Figure 1a). The longest contig was contig 3, with a length of 186,151,728 bp, and the shortest contig was contig 8, a double-bifurcation structure with a length of 1728 bp. Second, the Nanopore long reads resolved into double-bifurcation structures, with each contig merged using Bandage software (v0.8.1) (Figure 1b). Finally, one chromosome was obtained independently. The Nanopore long reads were then used to validate the T. wilfordii mitochondrial genome structure (Figure A2a–e).
The T. wilfordii mitochondrial genome was revealed to contain one chromosome 720,306 bp in length. The GC content was 45.54% (Figure 2), and it was revealed to be closely related to the Euonymus alatus mitochondrial genome in terms of size (genome size: 1,045,106 bp; NCBI accession number: NC_053921.1). In addition, the T. wilfordii mitochondrial genome was annotated as having a total of 61 genes (55 unique genes), including 35 PCGs, 22 tRNA genes (17 unique genes), and 4 rRNA genes (3 unique genes). The PCGs were classified into 10 functional groups (Table 2). The annotation of the core genes in the T. wilfordii mitochondrial genome was completed, revealing three variable gene types, including seven small subunit ribosome genes (rps1, rps10, rps12, rps13, rps19, rps3, and rps4), four large subunit ribosome genes (rpl10, rpl16, rpl2, and rpl5), and one subunit succinate dehydrogenase gene (sdh4).
2.2. Tandem Repeat Analysis
SSRs, also referred to as short tandem repeats or microsatellites, generally represent repetitive DNA sequences containing 1–6 nucleotides [15,16]. SSRs have been extensively used as genetic markers in forensics, disease diagnosis, and population genetics [17]. This work used the MISA online platform [18] for predicting the SSRs present in the T. wilfordii mitogenome. A total of 223 SSRs were identified on the chromosome of the mitogenome, including 66 monomers, 49 dimers, 29 trimers, 70 tetramers, 7 pentamers, and 2 hexamers (Table A2). These identified SSRs might be crucial molecular markers for subsequent analyses. LTRs, in which the repeat units contain ≥7 nucleotides, are crucial for numerous biological events, such as translation, transcription, promoter activity regulation, genome evolution, and chromosome stability [19,20,21]. The LTRs in the T. wilfordii mitochondrial genome were predicted using the Tandem Repeats Finder program [22] in this study. A total of 24 LTRs were predicted from the results (Table S1). LTR1 presented the greatest number of repeat units at 48 bp, whereas LTR16 was only 13 bp long.
2.3. Recombinations Mediated by Repetitive Sequences
Dispersed repetitive sequences (DRSs), or scattered repetitive sequences, are repeated at multiple positions across a genome and are used for determining biological evolution, individual identification, and disease diagnosis [23]. Furthermore, DRSs are crucial for determining mitogenome structural variations [24]. In this study, ROUS Finder 2.0.py [25] was used for predicting DRSs. A total of 47 DRSs were detected in the T. wilfordii mitochondrial genome, including 18 palindromic repetitive sequences and 29 direct repetitive sequences (Table 3). The DBSs were then compared to these DRSs, revealing four DBS sequences similar to DRS02–DRS05 (refer to Table 3, Figure S1, and Table S2). First, every repetitive sequence pair with 500 bp flanking sequences was isolated and used to create reference sequences representing the four possible conformations to be recombined. Second, the 47 DRSs were assessed for their recombination frequency with Nanopore long reads, revealing that no recombined conformation was supported by the Nanopore long reads (Table S2). Using the conformation as the baseline, it was determined that T. wilfordii has no four repetitive sequence-mediated recombinations.
2.4. Analysis of the Homologous Sequences Between Plastids and Mitochondria
Mitochondrial plastid DNAs (MTPTs) refer to plastid-derived DNA fragments in mitochondrial genomes [26]. These sequences originate from rare inter-organellar DNA transfer events, where fragments of the chloroplast genome are inadvertently integrated into the mitochondrial genome. Such transfers can cause misattribution of organelle DNA in taxonomic studies and complicate genome assembly/annotation, owing to chimeric origins. Therefore, the rigorous identification of MTPTs is essential for resolving these complexities. MTPTs have been reported in numerous species, such as Coffea arabica [27], Salvia miltiorrhiza [28], and Caragana spinosa [29]. In this study, the dataset adopted in the mitochondrial genome (OR538545.1/NC_082972.1) assembly was used for assembling the T. wilfordii chloroplast genome (OR538544.1), which is 156,692 bp long, with a GC content of 37.47%. The chloroplast genome included 131 genes (111 unique genes), comprising 87 PCGs (78 unique genes), 36 tRNA genes (29 unique genes), and 8 rRNA genes (4 unique genes) (Table S3 and Figure 3).
A total of eight MTPTs were identified in T. wilfordii by comparing the complete mitochondrial DNA with the chloroplast DNA from T. wilfordii using BLAST with default parameters [28]. These eight MTPTs were 3297 bp long, accounting for 0.46% of the entire mitochondrial genome. Among these, MTPT1 had the greatest length (1048 bp), whereas MTPT8 had the smallest length (30 bp). MTPT2 and MTPT7 were located in the inverted repeat (IR) region of the chloroplast genome (Figure S2). In addition, the locations of the eight MTPTs within the chloroplast and mitochondrial genomes are provided in Table S3 and Figure 3. In order to investigate the functions of these eight MTPTs, the DNA fragments were subjected to annotation, revealing that the MTPTs contained complete and functional ptDNA-encoded genes (trnW–CCA, trnP–UGG, trnS–GGA, and trnM–CAU) and partial-protein-encoding genes (psbD, psbC, and rpl2) (Figure 4 and Table S3). In order to further confirm that total MTPTs were present, Nanopore long reads were aligned to the reference sequences containing MTPTs as well as to the MTPT sequence and the corresponding 1000 bp upstream/downstream sequences. The mapping results revealed that the Nanopore long reads supported all eight MTPTs (Figure 4).
2.5. Phylogenetic Analysis
The NCBI nucleotide database has 136 mitochondrial genomes for Malpighiales, 2 for Celastrales, and 251 for Fabales. In order to analyze the phylogenetic relationships among the nine Celastrales and Malpighiales species, phylogenetic trees were created using the CDSs among the 24 common PCGs in the mitochondrial genomes (atp1, atp4, atp6, atp8, atp9, ccmB, ccmC, cox1, cox2, cox3, cob, matR, mttB, nad1, nad2, nad3, nad4, nad5, nad6, nad7,nad9, rps3, rps4, and rps12). It was found that T. wilfordii and Euonymus alatus were clustered under 100 bootstrap supports (Figure 5). These results showed that T. wilfordii and Euonymus alatus are more closely related. Additionally, each node showed a bootstrap support of >90, suggesting that these nine mitochondrial genomes of Celastrales and Malpighiales species have strongly reliable phylogenetic relationships.
2.6. RNA-Editing Event Analysis
RNA editing is an important supplement to the central dogma, and it differs from the corresponding DNA templates and exerts critical effects on various processes, including chloroplast and mitochondrial biogenesis, hormone and stress responses, and seed growth [29,30]. C-to-U conversion within plant organelles is the major RNA-editing pattern [30]. In this study, a total of RNA editing sites were identified in the T. wilfordii mitogenome following RNA-seq analysis (Table 4 and Figure S3). The false positives in the analysis of mitochondrial genomic polymorphic sites were eliminated by identifying the SNPs using the DNA-seq analysis of the identical samples of RNA-seq data. Three overlaps, cox2–698, cox2–721, and nad6–26, were obtained through the comparison of the predicted RNA-editing sites and SNP sites (Table S4 and Figure S3). Consequently, 600 RNA-editing sites were obtained from the T. wilfordii mitogenome (Table S4), among which 99 (16.50%) and 501 (83.50%) sites were altered, resulting in 99 synonymous and 501 non-synonymous codons, respectively. Variations in the number of non-synonymous codons for the Ser, Pro, and Arg amino acids were observed, among which 116 (19.33%) RNA-editing sites had Ser altered to Leu, 112 (8.67%) had Pro altered to Leu, 72 (12.00%) had Ser altered to Phe, 38 (6.33%) had Pro altered to Ser, 35 (5.83%) had Arg altered to Cys, and 34 (5.67%) had Arg altered to Trp (Table S4 and Figure 6a). The RNA-editing events within T. wilfordii mitochondria occurred mostly at the first and second codon base positions, accounting for 30.83% (185) and 52.67% (316) of the overall RNA-editing sites, respectively. Additionally, the RNA-editing events occurred in the most unique PCGs within the T. wilfordii mitochondrial genome. Among these genes, ccmB represented the highest number of RNA-editing sites (n = 45), followed by nad4 with 42 sites (Figure 6b). Notably, RNA-editing events resulted in the generation of both start and stop codons. Specifically, codon changes at positions atp6-718, atp9-223, and rps19-7 led to the conversion of CAA to TAA and CGA to TGA, effectively producing stop codons.
3. Discussion
3.1. Overview of the T. wilfordii Mitochondrial Genome
T. wilfordii is a traditional Chinese medicine that exhibits excellent medicinal effects in terms of curing nephrotic syndrome, rheumatoid arthritis, and systemic lupus erythematosus; in addition, this species has been extensively adopted in China as a folk medicine [31]. Acquiring the genomic data of this species constitutes a crucial step toward comprehending its active constituents’ physiological traits and biosynthetic processes. In this study, using hybrid assembly and annotation methods involving Illumina short reads and Oxford Nanopore long reads, the complete T. wilfordii mitogenome was identified. Subsequently, the gene contents and numbers, SSRs, TRSs, DRSs, homologous sequences, and RNA-editing events of the genome were determined. Finally, the phylogenetic relationships of the genome with eight Celastrales and Malpighiales species were explored according to the conserved PCGs in the mitochondrial genomes. The mitogenome of T. wilfordii will serve as a reference for investigating the genomes of other plants in the Celastraceae family and for Tripterygium mitogenome evolution and diversity research.
3.2. Architecture of One Molecule of the T. wilfordii Mitogenome
Plant mitogenome assembly is generally achieved and presented in circular maps according to the extensively accepted concept among living scientists that plant mitochondrial DNA exists mainly as molecules of circular genomes [32]. However, the physical structure of the real mitochondrial genome may involve different circles, complex branching structures, and linear molecules [33]. The results of this study revealed that the T. wilfordii mitochondrial genome is composed of one molecule that is 720,306 bp in length (Figure 1a,b). The molecular configuration, as depicted in Figure 1a, is characterized by the presence of two distinct bubble structures, one large and one small. In contrast, Figure 1b illustrates a linear structure. However, the linear structure is supported by the nanopore long reads (Figure A1 and Figure A2). The mitochondrial genomes of plants usually exhibit diverse alternative conformations because of the presence of many repetitive sequences [34,35]. According to this study, the T. wilfordii mitochondrial genome does not undergo recombination mediated by repetitive sequences. By extracting repetitive elements along with their 500 bp flanking regions as reference sequences and mapping them onto Nanopore long reads, it was confirmed that no recombination conformations associated with repetitive sequences are present in the T. wilfordii mitochondrial genome.
3.3. Research Trends for the T. wilfordii Mitogenome
The mitogenomes of plants differ significantly in length compared to chloroplast genomes due to frequent exchange with nuclear and chloroplast DNA [36,37]. In this study, the MTPTs of T. wilfordii were identified. These MTPTs were then subjected to functional annotation. MTPTs in the T. wilfordii mitochondrial genome included four complete and functional ptDNA-encoded genes (trnW–CCA, trnP–UGG, trnS–GGA, and trnM–CAU) and three partial-protein-coding genes (psbD, psbC, and rpl2). Furthermore, MTPT2 and MTPT7 were localized to the inverted repeat (IR) region of the chloroplast genome. Nonetheless, MTPT2, the partial rpl2, could be detected within the chloroplast and mitochondrial genomes. Thus, it was impossible to precisely determine whether MTPT3 originated from the T. wilfordii chloroplast or mitochondrial genome.
RNA editing, an extensive phenomenon observed in the organelles of higher plants, is used in several processes with different mechanisms that change the nucleotide sequences of RNA molecules so that the RNAs are different from the corresponding gene sequences [38,39]. In addition, RNA-editing events frequently occur at the first and second codon positions within the plant mitochondrial genome. In the T. wilfordii mitochondrial genome, this study identified 600 RNA-editing sites with alterations in one base, producing synonymous and non-synonymous amino acid codons, which in turn affected the translation of proteins. Ser, Pro, and Arg were the most frequently altered amino acids resulting from non-synonymous codon changes. Among the identified RNA-editing sites, only 99 (16.50%) occurred at the third codon position, while 501 (83.50%) were located at the first or second positions, indicating a predominant localization of editing events at the first and second codon positions. Furthermore, most PCGs in T. wilfordii contained RNA-editing sites, with the exception of rps13. These RNA-editing events can modify and refine genetic information, enhance the diversity of gene products, and thereby contribute to plant evolutionary adaptation.
4. Materials and Methods
4.1. Plant Materials, DNA and RNA Isolation, and Sequencing
Fresh T. wilfordii leaf samples were obtained from the nursery of the medical school of Huaqiao University (Quanzhou, China). Thereafter, a DNA extraction kit and an RNAprep Pure Plant Plus Kit (Tiangen Biotech, Beijing, China) were used to extract the total DNA and RNA, respectively, which were preserved at −80 °C until use. Specifically, the extracted total DNA was used for Illumina sequencing and Oxford Nanopore sequencing (Table A1). For Illumina sequencing, 1 µg of the genomic DNA was used to construct a DNA library with the NEBNext library building kit, followed by Illumina 2500 platform sequencing (Illumina, San Diego, CA, USA). For Oxford Nanopore sequencing, a 10 kb DNA library was established prior to sequencing using a PromethION sequencer (Oxford Nanopore Technologies plc, Oxford, UK). The TruSeq Stranded mRNA Library Prep kit (Illumina) was used to construct a strand-specific RNA-seq library. Illumina sequencing was performed using PE150 sequencing for the quantified library.
4.2. Genome Assembly, Annotation, and Validation
GetOrganelle software (3.8.2)was used to complete the organelle genome assembly [40] In this study, the parameters “–R 15–k 21,45,65,85,105–F embplant_pt” were adopted for assembling the chloroplast genome. However, mitochondria-derived nuclear and plastid DNA can result in false positives during mitogenome polishing [41]. Thus, mitochondrial genome assembly was completed using the single hybrid assembly strategy. First, the parameters “–R 20 –k 21,45,65,85,105 –P 1000,000 –F embplant_mt” were used to obtain mitochondrial short reads using GetOrganelle (3.8.2) [40]. Second, long and short reads were assembled through de novo assembly into a unitig graph using SPAdes [42], miniasm, and Racon packages [43] included in Unicycler software (v0.5.0). Finally, using Unicycler software, the Nanopore long reads were adopted to address the double-bifurcating structures (DBSs) in the unitig graph [44]. The bandage software was applied to visualize the connections of contigs to allow for the manual removal of nodes derived from the chloroplasts and nuclei [45]. CPGAVAS2(v2.0) [46] was used for chloroplast genome annotation, whereas the CPGView(v1.0) web server [47] was used for potential annotation errors in the chloroplast genome”. However, the annotation of the mitogenome was completed using the GeSeq [48] and PMGA (http://www.1kmpg.cn/mgavas/, accessed on 14 July 2025) web servers. Thereafter, Apollo software (v1.11.8) [49] was used for the manual correction of the annotation results. The chloroplast and mitochondrial genome structures were subsequently drawn with PMGmap (http://www.1kmpg.cn/pmgmap, accessed on 14 July 2025). Finally, the organelle genome sequences and the annotations were deposited into GenBank (accession nos. OR538544.1 and OR538545.1 (NC_082972.1) for the chloroplast and mitochondrial genomes, respectively) [50].
4.3. Identification of Tandem Sequences
MISA [18] and Tandem Repeats Finder [22] are two web servers that are used to analyze simple sequence repeats (SSRs) and long tandem repeats (LTRs), respectively. SSRs, also referred to as short tandem repeats or microsatellites, were analyzed with MISA, using the thresholds of below 10, 5, 4, and 3 units for mononucleotides, dinucleotides, trinucleotides, and tetra-/penta-/hexa-nucleotides, respectively. Moreover, LTRs were predicted with Tandem Repeats Finder using the default parameters of 7 and 2 for mismatches/indels, which were matched, respectively, with a maximal period size of 500 and a minimal alignment score of 50.
4.4. Identification of Repeat-Mediated Recombination
In order to identify repeat-mediated recombination, the dispersed repeat sequences of the mitochondrial genome were detected with ROUS Finder 2.0.py [25]. Thereafter, the repetitive sequences and their bilateral flanking sequences (500 bp) were extracted; these two sequences corresponded to one conformation. Later, the two sequences were integrated in silico to generate the sequences associated with the other conformations that were recombined. Next, the Nanopore long reads were mapped to four DNA sequences in two conformations, followed by counting the number of mapped reads with BWA (v0.7.17) [51] and SAMtools (v1.17) [52]. Finally, IGV software (v.2.8) was adopted to visualize the read mapping results [53]. In order to further confirm the T. wilfordii mitochondrial genome structure, the initial and terminal 1000 bp sequences of the genome were extracted and concatenated. Next, 1000 bp sequences were extracted and concatenated from the junction of contig10 and contig11, the junction of contig6 and contig12, and the junction of contig14 in the Tripterygium wilfordii mitochondrial genome. Thereafter, the Nanopore long reads were mapped to these DNA sequences using BWA (v0.7.17) [51] and SAMtools (v1.17) [52]. Finally, IGV software (v.2.8) [53] was adopted to visualize the read mapping results and verify the T. wilfordii mitochondrial genome structure.
4.5. Identification of Mitochondrial Plastid Sequences (MTPTs)
Plastid genomes are transferred to plant mitogenomes to generate MTPTs, which can influence the complexity of mitochondrial genomes and can induce the false-positive DNA barcoding paradox [54,55]. Therefore, MTPTs were identified in this study by comparing the cpgenome (OR538544.1) with the mitogenome (OR53 8545.1/NC_08 2972.1) of T. wilfordii using BLASTN software (v2.2.30+) [56] with default parameters. In order to confirm the identified MTPTs, the MTPT sequence and its upstream/downstream sequences (1000 bp) were obtained to create the reference genome. Thereafter, using default parameters, the Nanopore long reads were aligned to these reference sequences with BWA software (v0.7.17) [52]. Finally, IGV software (v.2.8 ) was used to visualize the Nanopore long-read mapping results to the MTPT regions [53]. The Circos package of TB tools (V1.098) was then applied to visualize the MTPT results [57,58].
4.6. Phylogenetic Tree Construction
A total of 9 mitochondrial genomes of Celastrales and Malpighiales species were used in the phylogenetic analysis, and Caragana spinosa (OQ785640.1) was selected as the out group. The following mitochondrial genomes were downloaded from the NCBI GenBank database: Tripterygium wilfordii (OR538545.1/NC_082972.1), Euonymus alatus (N C_053921.1), Populusdavidiana (NC_035157.1), Populusrotundifolia (MW566588.1), Salixsu chowensis (NC_029317.1), Passiflora edulis (NC_050950.1), Hevea brasiliensis (AP014526.1), Manihot esculenta (NC_0451 36.1), and Jatropha curcas (OQ603497.1). For phylogenetic analysis, the common coding sequences (CDSs) among the afore-stated species were obtained with PhyloSuite software3 (v.1.2.1) [59] and then aligned using MAFFT software4 (v7.505) [60]. Using the maximum likelihood approach, the aligned sequences were subsequently used to construct a phylogenetic tree with IQ-Tree(v2.1.4-beta) [61]. UFBoot was then used for performing bootstrap analysis involving 1000 replicates [62]. Finally, iTOL (https://itol.embl.de/, accessed on 14 July 2025) was used for visualizing the resulting phylogenetic tree [63].
4.7. RNA-Editing Site Identification in the PCGs of the T. wilfordii Mitogenome
In this study, to identify the RNA-editing sites within the T. wilfordii mitogenome, strand-specific RNA-seq data were mapped to 100 bp of the sequences in 35 PCGs and the corresponding 3′and 5′ flanking regions using BWA (Burrows–Wheeler Alignment tool) [51]; moreover, reference sequence-matching alignment reads were obtained with SAMtools (v1.17) [52]. Thereafter, REDItools [64] was used to predict the RNA-editing sites using specific parameters of frequency ≥ 0.1 and coverage ≥ 5 [15]. In order to increase the prediction accuracy, the RNA-editing sites were visualized using IGV software (v.2.8 ) [53], with a special focus on sites with frequency > 0.2. In order to exclude the impact of single-nucleotide polymorphism (SNP) sites on the findings, SNPs were also predicted, and RNA-editing sites overlapping with the results of the SNP analysis were eliminated. Similarly, when the SNPs of the T. wilfordii mitogenome were identified, the Illumina short reads in the whole genome were mapped to 100 base pairs of CDSs in the PCGs, the corresponding 3′and 5′ flanking regions were mapped with BWA software (v0.7.17) [51], and isolated alignment reads matching the reference sequence were mapped with SAM tools (v1.17) [52]. Subsequently, the SNP sites were analyzed using REDItools (http://code.google.com/p/reditools/m accessed on 14 July 2025) [64], with parameters requiring frequency ≥ 0.1 and coverage ≥ 5.
5. Conclusions
This study is the first to report the T. wilfordii mitochondrial genome according to the sequences obtained from Illumina short reads and Nanopore long reads. The T. wilfordii mitogenome is a unique chromosome. This study revealed the general features of the T. wilfordii mitochondrial genome through various comparative analyses, including repetitive sequence analysis, recombination analysis, homologous sequence analysis, phylogenetic analysis, and RNA-editing event analysis. Furthermore, it was found that the T. wilfordii mitochondrial genome exhibits no recombination conformation mediated by repetitive sequences.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Zhong Y. Zhang J.Z. Bao Z.Z. The complete chloroplast genome of Tripterygium wilfordii Hook. f.(Celastraceae)Mitochondrial DNA Part B 202271696169810.1080/23802359.2022.211910336188665 PMC 9518234 · doi ↗ · pubmed ↗
- 2Liu Y.F. Tu S.H. Gao W.N. Wang Y. Liu P. Hu Y.H. Dong H. Extracts of Tripterygium wilfordii Hook F in the treatment of rheumatoid arthritis: A systemic review and meta-analysis of randomised controlled trials Evid.-Based Complement. Altern. Med.201320134107910.1155/2013/41079324391674 PMC 3866793 · doi ↗ · pubmed ↗
- 3Chen F.Y. Liu J.T. Zhao Z.M. Li Z.P. Wu K.Y. Tripterygium and its plant extraction for systemic lupus erythematosus: A protocol for systematic review and meta analysis Medicine 202099 e 2190910.1097/MD.000000000002190932846857 PMC 7447359 · doi ↗ · pubmed ↗
- 4Chen Y.W. Lin G.J. Chia W.T. Lin C.K. Chuang Y.P. Sytwu H.K. Triptolide exerts anti-tumor effect on oral cancer and KB cells in vitro and in vivo Oral Oncol.20094556256810.1016/j.oraloncology.2008.10.00719359213 · doi ↗ · pubmed ↗
- 5Kuchta K. Xiang Y. Huang S. Tang Y. Peng X. Wang X. Zhu Y. Li J. Xu J. Lin Z. Celastrol, an active constituent of the TCM plant Tripterygium wilfordii Hook. f., inhibits prostate cancer bone metastasis Prostate Cancer Prostatic Dis.20172015616410.1038/pcan.2016.6128195223 · doi ↗ · pubmed ↗
- 6Li J.H. Hao J.J. Treatment of neurodegenerative diseases with bioactive components of Tripterygium wilfordii Am. J. Chin. Med.20194776978510.1142/S 0192415 X 1950040 X 31091976 · doi ↗ · pubmed ↗
- 7Yang C. Su C. Iyaswamy A. Krishnamoorthi S.K. Zhu Z. Yang S. Tong B.C. Liu J. Sreenivasmurthy S.G. Guan X. Celastrol enhances transcription factor EB (TFEB)-mediated autophagy and mitigates Tau pathology: Implications for Alzheimer’s disease therapy Acta Pharm. Sin. B 2022121707172210.1016/j.apsb.2022.01.01735847498 PMC 9279716 · doi ↗ · pubmed ↗
- 8Xu S.H. Feng Y.Q. He W.S. Xu W. Xu W. Yang H.J. Li X.Y. Celastrol in metabolic diseases: Progress and application prospects Pharmacol. Res.202116710557210.1016/j.phrs.2021.10557233753246 · doi ↗ · pubmed ↗