Unraveling the Role of Autotaxin and Lysophosphatidic Acid in Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Potential
Jesús García-de Soto, Mónica Castro-Mosquera, Jessica María Pouso-Diz, Alejandro Fernández-Cabrera, Mariña Rodríguez-Arrizabalaga, Manuel Debasa-Mouce, Javier Camino-Castiñeiras, Anxo Manuel Minguillón Pereiro, Marta Aramburu-Núñez, Daniel Romaus-Sanjurjo, José Manuel Aldrey

TL;DR
This paper reviews how autotaxin and lysophosphatidic acid may contribute to Alzheimer's disease and could be used as new treatment targets.
Contribution
The paper provides a synthesis of recent findings on the role of the ATX/LPA axis in Alzheimer's disease.
Findings
ATX and LPA are involved in neuroinflammation and blood–brain barrier dysfunction in Alzheimer's disease.
LPA signaling affects synaptic plasticity and tau pathology, contributing to AD progression.
The ATX/LPA axis shows potential as a biomarker and therapeutic target for Alzheimer's disease.
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by the accumulation of amyloid-β plaques, tau hyperphosphorylation, and chronic neuroinflammation. Emerging evidence suggests a crucial role of lipid signaling pathways in AD pathogenesis, particularly those mediated by autotaxin (ATX) and lysophosphatidic acid (LPA). ATX, an enzyme responsible for LPA production, has been implicated in neuroinflammatory processes, blood–brain barrier dysfunction, and neuronal degeneration. LPA signaling, through its interaction with specific G-protein-coupled receptors, influences neuroinflammation, synaptic plasticity, and tau pathology, all of which contribute to AD progression. This review synthesizes recent findings on the ATX/LPA axis in AD, exploring its potential as a biomarker and therapeutic target. Understanding the mechanistic links between ATX, LPA, and AD…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
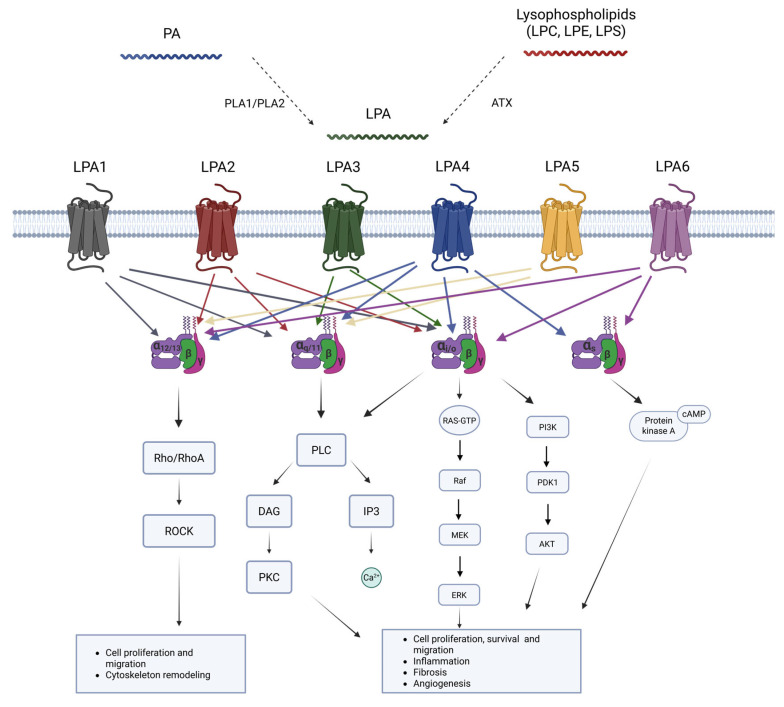 Figure 1
Figure 1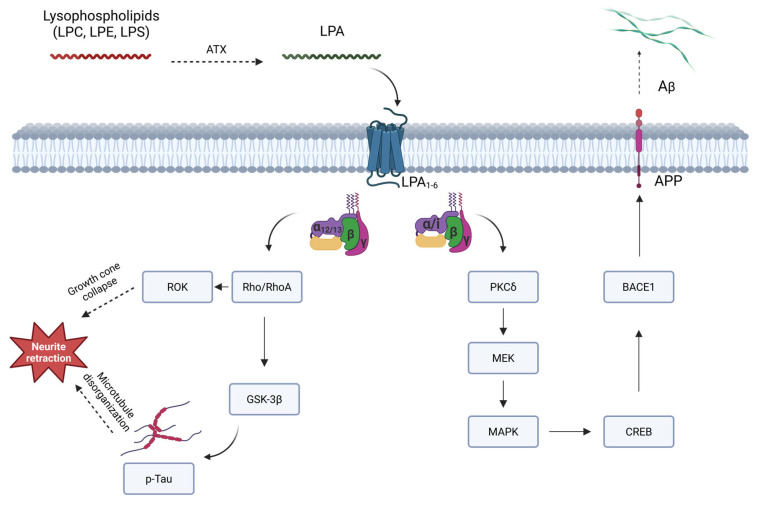 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSphingolipid Metabolism and Signaling · Endoplasmic Reticulum Stress and Disease · Alzheimer's disease research and treatments
1. Introduction
Alzheimer’s disease (AD) is the leading neurodegenerative cause of dementia in developed countries [1]. In the United States, it is estimated that 6.2 million individuals aged 65 and older are living with AD, a number that could reach 13.8 million by 2060 [1]. AD ranks as the sixth-leading cause of death in the US [1]. Clinical manifestations of AD encompass cognitive impairment, psychiatric symptoms, behavioral changes, sleep disturbances, movement disorders, and seizures [2]. It invariably leads to functional impairment, as no treatment has been proven to cure the disease [2].
The major neuropathological features of AD include extracellular deposits of β-amyloid (Aβ), which form amyloid plaques and intracellular accumulation of hyperphosphorylated tau protein in neurofibrillary tangles (NFT) [3]. Additionally, AD has been associated with vascular pathology [4], neuroinflammation [5], oxidative stress [6], and alterations in lipidic metabolism [7,8].
Lipid metabolism has been extensively studied and established as a significant risk factor for the development of AD [9,10,11]. The primary genetic risk factor for developing late-onset AD is the presence of the e4 allele of the apolipoprotein E gene (APOE4). Among the various isoforms of APOE, APOE3 does not appear to influence AD, APOE2 (the least common) may have a protective effect, while APOE4 increases the risk of developing the disease [12,13,14].
The brain is composed of lipids in approximately 60% to 70% by dry weight, which are mainly structural components. However, many of them have cellular signaling properties. In this regard, lysophospholipids are metabolic intermediates derived from membrane phospholipids via hydrolysis. Interestingly, intracellular lysophospholipids are intermediate precursors for the biosynthesis of other lipids, while extracellular lysophospholipids act as signaling molecules. In recent years, lysophosphatidic acid (LPA), one of the most important lysophospholipids, has been studied as a key modulator in several diseases [15].
LPA is a bioactive lipid that acts as a potent extracellular signaling molecule with numerous systemic functions [16], including its involvement in central nervous system (CNS) development. The implication of LPA in brain development is well known [15,17]. Therefore, dysregulation of LPA has been suggested to be involved in several neurological disorders [16,18]. Autotaxin (ATX) is an extracellular enzyme responsible for converting lysophospholipids into LPA, maintaining physiological concentrations of the latter, thus guaranteeing its function as a signaling molecule [18]. In this regard, in recent years the involvement of the ATX/LPA axis has been observed in different neurological diseases, such as AD [19], Parkinson’s disease [20], major depressive disorder [21], neuropathic pain [22], migraine [23], and loss of blood–brain barrier (BBB) integrity [24].
This review explores the role of ATX and LPA signaling in the pathogenesis of AD, highlighting their involvement in neuroinflammation, tau phosphorylation, and β-amyloid metabolism.
2. Autotaxin and Lysophosphatidic Acid
ATX, also known as ectonucleotide pyrophosphatase/phosphodiesterase (ENPP2, encoded by the homonym gene), is a secreted lysophospholipase D (lysoPLD) that belongs to the ENPP family [25]. ATX is widely expressed, with the highest mRNA levels detected in the brain, spinal cord, ovaries, lungs, intestines, and kidneys [24].
ATX is synthesized as a preproenzyme requiring post-transcriptional modifications to be active [25]. The ENPP2 gene undergoes alternative splicing and produces four known isoforms (α, β, γ, and δ). ATX β is the canonical, predominant, and most studied form. Interestingly, ATX γ is a “brain-specific” isoform. However, despite the differences in structure, it is currently unclear whether the isoforms are associated with distinct physiopathological conditions or not [25].
LPA (1- or 2-acyl-sn-glycerol 3-phosphate/radyl-glycerol-phosphate), consists of a glycerol backbone, a phosphate group at the sn-3 position, and a hydroxyl group added to a fatty acid chain in sn-1 or sn-2 positions. Different variants are formed depending on the acyl chain length, with 16:0-LPA, 18:2-LPA, and 18:1-LPA being the most abundant in humans. In this regard, the 18:1-LPA form is commonly used in research [16].
ATX primarily produces LPA from extracellular lysophosphatidylcholine (LPC), although it can also degrade lysophosphatidylethanolamine and lysophosphatidylserine with less affinity [26,27]. It is well established that ATX is the main source of synthesized LPA [28]. Due to rapid clearance from circulation through liver degradation [29], the main biological effects of the ATX/LPA axis result from LPA signaling. Extracellular LPA can be found in various biological fluids, such as plasma, cerebrospinal fluid (CSF), saliva, tears, and aqueous humor [26,30]. The highest serum levels of LPA are produced by platelets during clotting [31], when concentrations of 10–15 μM in serum can be reached [30]. In this context, concentrations of less than 1 μM in plasma are considered “normal”; and even lower levels in cerebrospinal fluid (CSF) [30].
LPA has been shown to play a role in numerous physiological functions, including blood pressure regulation, cell proliferation and survival, calcium mobility, and angiogenesis, among others [32]. It has also been implicated in pathological processes, such as different types of cancer [33,34,35,36,37,38,39,40,41], rheumatologic diseases [42,43], pulmonary fibrosis [44,45], psychiatric disorders, and neurological disorders [23,46,47,48,49].
In addition to extracellular production, there is also intracellular production of LPA through pathways including monoacylglycerol kinase (MAGK), PA-PLA1 or PA-PLA2, glycerophosphate acyltransferase (GPAT) synthesis, and the oxidative modification of low-density lipoprotein (LDL) [16]. Intracellular LPA functions as a substrate for glycerolipid synthesis and is unlikely to serve as an extracellular mediator [18].
3. LPA Signaling in the Central Nervous System
LPA acts through six specific G protein-coupled receptors (GPCRs) to maintain homeostasis in relevant processes such as cell proliferation, migration, or cytoskeletal reorganization [16,30,50,51]. These six identified receptors (LPA1 to LPA6) [52] differ in their G-protein coupling, downstream signaling pathways, tissue distribution, and physiological functions (Table 1). They are also encoded by different genes (LPAR1–6) [53].
The first three LPA receptors (LPA1–3) belong to the endothelial differentiation gene (Edg) subfamily and have been studied in more detail, whereas the last three (LPA4–6) have been more recently discovered, and belong to the phylogenetically distant non-Edg subfamily of receptors [54]. All these receptors are 7-transmembrane GPCRs and exhibit high affinity for LPA. Consequently, these GPCRs are coupled with one or several heterotrimeric G-proteins from the four existing classes (Gα12/13, Gαq/11, Gαi/o, and Gαs), which can initiate a wide range of downstream pathways (Table 1) [52].
As mentioned, LPA receptors are widely distributed throughout several systems in the organism, accounting for their equally diverse biological functions. Among these systems, the CNS stands out for the greater significance of LPA signaling [55,56]. LPA, acting through its receptors LPA1–6, has a main role in the early stages of mouse brain development and neurogenesis [57,58,59]. Moreover, LPA and its analogs have been observed to promote neurotrophic effects and neurite outgrowth in cultured cells [60]. Furthermore, LPA has been suggested to play a role in synaptic plasticity and modulation, and it could also be involved in the development of neuropathic pain [61]. LPA receptors are also found in CNS glial cells, where they play a role in mediating the development of oligodendrocytes, activating microglia and potentially inducing neuronal differentiation [17,32,51,55,62]. Furthermore, these receptors may have a regulatory function in activating microglia-mediated neuroinflammation through mitogen-activated protein kinase (MAPK) and AKT activation [63]. A summary of LPA metabolism and its receptors can be found in Figure 1.
In addition to its physiological functions, LPA signaling is involved in the pathogenesis of diseases, including neurological diseases (such as AD, Parkinson’s disease, and Huntington’s disease) [64,65], heart and vascular conditions (for instance, heart failure) [66,67], respiratory pathologies (i.e., pulmonary fibrosis or pulmonary hypertension [68]), gastrointestinal afflictions [69], and different types of cancer [70]. Under pathological conditions, LPA signaling can undergo significant alterations, thus contributing to disease progression [30]. Different LPA receptors mediate pathogenesis-related processes in the CNS in several ways, as summarized in Table 2. Therefore, studying them as potential biomarkers or therapeutic targets proves to be a promising strategy in the search for new drugs or diagnostic tests.
3.1. LPA1
LPA1, being the first receptor to be discovered [100,101], is also the most extensively studied. As with LPA2 and LPA3, it belongs to the family of the endothelial differentiation gene (Edg) receptors. Initially named Vzg-1, this receptor was isolated from the ventricular zone of a neuroblast cell line, where it is highly expressed [100]. LPA1 is the most prevalent receptor in human and mouse brain tissue, and it can be coupled with three of the Gα proteins, Gαi/o, Gαq/11, and Gα12/13, thereby initiating downstream signaling cascades through phospholipase C, MAPK, protein kinase B (PKB, also known as Akt), and Rho [30]. Recently, the LPA1 structure was elucidated, providing new insights into its activation mechanisms [102]. Its K_d_ values vary, depending on the LPA species, between 0.87 and 69 nM [103,104,105]. Beyond the CNS, LPA1 presents a broad distribution, promoting a diverse range of cellular responses, including cell proliferation and survival, and Ca^2+^ mobilization, among others [32,106].
Extensive research has been conducted concerning the role of LPA1 in neurodevelopment, as it participates in a wide range of processes. Some notable examples include its involvement in regulating myelination during the postnatal period, as well as in developing white matter tracks [107,108], in cortical development [109,110,111], and in cell migration [112]. Additionally, LPA1 plays an essential role in the development of the peripheral nervous system by controlling the function and growth of Schwann cells [113]. In this regard, mutant mouse models lacking this specific receptor show a significant impairment in hippocampal neurogenesis [114]. Moreover, descendants of the Lpar1−/− line have only a 50% survival rate due to impaired suckling behavior, and the surviving half display anomalies such as reduced body size, cranial dysmorphism, and increased apoptosis in the Schwann cell population of the sciatic nerve [115,116]. In recent years, new knock-in mouse models have been developed, replacing one of the wild-type alleles of LPA1 with a new mutant (Lpar1-EGFP and LPA1-LacZ). This has enabled new in situ studies of this receptor’s functions, showing that LPA1 is highly expressed in oligodendrocytes [117,118]. Overall, the evidence gathered strongly suggests that LPA1 is a crucial modulator of neurogenesis.
LPA1 has also been found to play a role in neuropathic pain [22]. LPA1 and LPA3 receptor-deficient mice showed absent allodynia in paclitaxel-induced neuropathy [119], neuropathic pain due to diabetic neuropathy [120], and central post-stroke pain [121]. Accordingly, mice treated with LPA1 and LPA3 antagonists showed reversed abnormal pain behaviors [119,120,121].
3.2. LPA2
LPA2 is another well-characterized receptor [122] that promotes neurogenesis by inducing neurite formation, like LPA1 [123], with some overlapping functions. Its K_d_ is approximately 64 nM [105]. LPA2 exhibits a more diffuse expression pattern in the developing brain [30,124]. However, LPA2 null mice do not exhibit any obvious differences in phenotype [125], in contrast to those lacking LPA1, which might suggest a redundant effect of LPA2 in the developing mouse brain. The lack of both receptors in double-knockout mice showed reduced LPA-induced responses in embryonic fibroblasts [125].
Interestingly, it has been reported that an upregulation of LPA2 exists in the adult mouse brain after traumatic brain injury (TBI) in the astrocytes and neurons [126,127]. This upregulation has also been observed in astrocytes after spinal cord injury [127], as well as after ischemia in the inner layers of rat retina, suggesting compensatory mechanisms to promote cell survival [128].
3.3. LPA3
LPA3 was initially studied for its role in embryo implantation [129]. In recent years, it has also been studied as a potential biomarker and therapeutic target in ovarian cancer [130].
LPA3 plays an essential role in the formation of neuronal networks [131], as it can mediate axonal branching (in contrast to other LPA receptors), and it also plays a critical role regulating the expression of the antioxidant enzymes that eliminate reactive oxygen species (ROS) [132]. Likewise, it has been found to increase its expression following brain injury [127]. More recently, a highly specific LPA3 agonist, ADS024-IPA, has been shown to improve outcomes when given orally to different animal models of neuroinflammatory disease, suggesting a potential role as a prophylaxis or treatment for these pathologies [133]. LPA is a poor ligand for LPA3 [134].
As previously mentioned, LPA3 may also participate in the regulation of neuropathic pain, like LPA1, and treatment with LPA1 and LPA3 antagonists was useful in mice as a potential chronic pain treatment [22,119,120,121].
3.4. LPA4
LPA4 was the first receptor to be characterized with a structural difference from the previously discovered LPA receptors, as it is a non-Edg receptor [54]. LPA4 has a specific binding affinity to 18:1-LPA with a K_d_ value of 44.8 nM to 100 nM, but not to other lysophospholipids and related lipids, such as S1P and SPC [104,105,135].
This receptor can induce neurite retraction and stress fiber formation through the Rho/ROCK pathways [30,136,137]. Intracellularly, LPA4 increases concentrations of Ca^2+^ and cAMP, the latter serving as a complementary effect against the cAMP attenuating activities of LPA1–3 [137]. LPA4 also mediates ROCK-dependent cell aggregation and N-cadherin-dependent cell adhesion [136]. It is noteworthy that LPA4 negatively regulates cell motility and inhibits the effects of LPA1 on cell migration [138]. The LPA4 axis is required for bipolar morphogenesis and radial migration, modifying the actin cytoskeleton of newborn cortical neurons [139]. Interestingly, LPA4 null mouse lines appear mostly normal [138], like LPA2 null lines.
3.5. LPA5
Discovered in 2006, LPA5 is another non-Edg LPA receptor, which binds to Gαq and Gα12/13, Gαi, and Gαs [16,30,140]. Its K_d_ ranges from 6 to 89 nM [104,105,140]. It is involved in neurite retraction mediated by Gα12/13 and Rho [137].
Additionally, LPA5 is speculated to be involved in nociception and pain hypersensitivity mechanisms, as well as in anxiety-related and motivation-driven behaviors [141]. Moreover, it is highly expressed in the dorsal root ganglia [140], thus suggesting its possible involvement in neuropathic pain. This was confirmed in Lpar5 null mice, which had decreased pain and a lower level of phosphorylated cyclic AMP response element binding protein (pCREB) expression [95]. The use of the LPA5 antagonist AS2717637 in rodents has also shown analgesic effects [142]. The use of another LPA5 antagonist, TCLPA5, has been shown to disrupt the pro-inflammatory effects of this receptor, interfering with microglia polarization [143].
3.6. LPA6
LPA6, the most recently discovered receptor, is predominantly expressed in brain capillary endothelial cells, where the LPA–LPA6–G12/13–Rho pathway plays a part in BBB permeability [98]. Nevertheless, much remains unknown about the role of this receptor in the CNS. Its K_d_ has not been accurately determined yet, probably due to low affinity or the rapid off rates of LPA [105]. In addition, a recent study demonstrated that LPA6 expression in oligodendrocytes could be considered a negative regulator of myelination during CNS development [99].
4. ATX/LPA Axis and Alzheimer’s Disease
It is well known that the accumulation of Aβ is the hallmark of AD [144]. The increasing deposits of Aβ produce neurotoxicity in the CNS [145], contributing to tau hyperphosphorylation [146], and resulting in neuronal cell death. Recently, a clear correlation between dysfunctional LPA signaling and the pathogenesis of AD has been established [19], along with alterations in other bioactive lipids, such as S1P [8,62].
Furthermore, substantial evidence supports a significant degree of oxidative damage to lipids and proteins throughout the progression of AD [147]. CSF lipoproteins are particularly sensitive to oxidation, becoming neurotoxic in their oxidized state [148]. Interestingly, LPA is the main bioactive component of oxidized low-density lipoprotein (oxLDL), a high-cardiovascular risk molecule [149]. Regarding AD, oxLDL-LPA induces an elevation in Aβ production by activating the LPA/BACE1/APP axis [150].
LPA can induce the activation of delta protein kinase C (PKCδ) [151], triggering a downstream signaling cascade through consecutive phosphorylation of mitogen-activated protein kinase kinase (MEK), MAPK, p90RSK, and finally cyclic AMP response element binding protein (CREB) [150]—a transcription factor known to upregulate BACE1 expression by binding to its gene promoter region when active [152]. BACE1 [153], also known as beta-site amyloid precursor protein cleaving enzyme 1 or simply beta-secretase, together with gamma-secretase [154], are the secretases directly responsible for producing Aβ from amyloid precursor protein (APP) [155].
Cholesterol is another lipidic molecule that has been linked to the pathogenesis of AD, also through its contribution to the accumulation of Aβ [156]. Lipid rafts, domains in the plasma membrane [157] enriched in cholesterol and sphingolipids, play a role in promoting the interaction between APP and BACE1 [158]. OxLDL is one of the major cholesterol carriers and contributes to the increase in lipid raft formation along the plasma membrane in AD through a redox-imbalance process [159]. The oxidative stress caused by oxLDL triggers glutathione (GSH) peroxidase activity, leading to the depletion of GSH [160]. In the absence of GSH, the enzyme sphingomyelinase increases its activity, likewise contributing to lipid raft formation, and consequently increasing Aβ formation [159].
As mentioned, post-translational modifications of tau are also key mediators in the pathogenesis of AD [146]. Tau is an essential component for the proper functioning of microtubules [161]. Hyperphosphorylated tau forms aggregates of paired helical filaments, and the extreme aggregation of these filaments results in the neurofibrillary tangles responsible for the pathogenesis of AD [162]. In this sense, LPA can contribute to tau phosphorylation through the action of glycogen synthase kinase-3β (GSK-3β) [163,164]. This kinase is highly expressed in the brain and, under normal conditions, plays a role in axonal growth during neurodevelopment [165]. However, dysregulations of this enzyme are associated with the vast majority of pathological processes in AD [166]. The most probable mechanism involves the LPA/RhoA/ROCK pathway, as observed in other related cases [167]. In fact, it has already been documented that Rho kinases are involved in tau hyperphosphorylation [168]. Furthermore, GSK-3β inhibitors have been shown to block tau hyperphosphorylation and to improve tauopathies [169], confirming the involvement of this enzyme in the process. These mechanisms are represented in Figure 2.
4.1. ATX and LPA as Potential Biomarkers in Alzheimer’s Disease
As mentioned earlier, LPA mediates neurite retraction during neuronal development. However, anomalies can result in an excessive amount of neurite retraction, leading to diseases in adult individuals [164]. LPA-induced tau hyperphosphorylation by GSK-3β is a potential cause of pathological neurite retraction and is linked with AD [170]. While inhibitors of GSK-3β improve neurite retraction, they do not block it completely [163], indicating the involvement of other important factors. For example, kinases, such as p38 MAPK [171] and CDK5 [172], are also known to be linked to this process.
These interactions are further supported by the research on LPA levels in CSF. Various isoforms of this lysophospholipid were analyzed in CSF, demonstrating a significant association between their levels and concentrations of classical AD biomarkers, such as Aβ-42, p-tau, and total tau [173].
LPA1 is also highly relevant in the context of AD. As previously mentioned, various models of LPA1-null mice have provided insights into the functions of this receptor. Behavioral studies have indicated that, in the absence of LPA1, mice exhibit anxiety, memory impairment, and motor abnormalities [174,175], highlighting the role of this receptor in the correct functioning of these processes. An expression profiling of circular RNAs in CSF from AD patients revealed an upregulation of Circ-LPAR1 expression, suggesting its potential as a biomarker for AD [176]. Moreover, Circ-LPAR1 has been suggested to be involved in inducing neuronal apoptosis, inflammation, and oxidative stress [177].
In a recent study [178] carried out on a triple transgenic mouse model of AD, LPA1 activity was found to be increased in the corpus callosum, motor cortex, hippocampal CA1 area, and striatum. The authors postulated that this increase could be related to adaptations during the development of the 3xTg-AD mice, increasing the demand for the LPA endogenous neurotransmitter and elevating the levels of lipid precursors.
Moreover, ATX is also associated with AD. Studies have observed significantly higher expression of ATX in the frontal cortex of patients suffering from AD [179]. ATX concentrations in CSF can serve as a biomarker of brain metabolic dysfunction, as they have been found to be increased in patients with AD and mild cognitive impairment (MCI). This elevation correlates with hypometabolism in the prefrontal cortex and mesial temporal lobes in positron emission tomography (PET) scans, as well as with concentrations of classical AD biomarkers [180]. However, further investigation into the underlying mechanisms is needed.
Another known risk factor for AD is TBI [181]. TBI causes BBB dysfunction [182], leading to an increased presence of LPA in the CNS. Therefore, there is an amplification of all the downstream consequences mentioned above, creating a positive feedback loop, as high concentrations of LPA also contribute to the permeabilization of the BBB through the Rho/ROCK/MMP-9 or uPA pathways [183].
4.2. Advances and Challenges in Therapy Targeting the ATX/LPA Axis
In addition to its roles earlier discussed, LPA1 serves as an important mediator in adult hippocampal neurogenesis [184], the hippocampus being a key region for memory, and therefore highly relevant in the context of AD [185]. Studies have shown that LPA administration can decrease cocaine-contextual memory in models of cocaine addiction by stimulating hippocampal neurogenesis [184]. Further research is needed on the topic of LPA-induced adult hippocampal neurogenesis to explore potential applications for the development of new strategies against AD. Indeed, the creation of new therapies involving LPA1 and other receptors may not only serve as valuable tools for functional studies but may open avenues for the design of new treatments [186,187,188,189].
As mentioned earlier, the hippocampus is a key region for memory. Pathological concentrations of LPA can induce apoptosis/necrosis of the hippocampal neurons [190]. However, LPA is also necessary for the proper functioning of the hippocampus. Inhibitors of LPA receptors have shown that LPA regulates cognition and emotion [191]. Also, the administration of external LPA [191] or antidepressants that behave as agonists of LPA receptors [192] can act as potential treatments favoring the neuroprotection of the hippocampus.
Neuroinflammation has neurotoxic effects on the CNS and is closely related to the pathogenesis of AD [5]. Markers associated with the immune system have been found in the brain and biological fluids of patients with this disorder [193,194]. Microglial activation correlates with NFT and amyloid plaque load in AD and MCI [195]. More recently, the inflammasome has also been suggested to play a role in the disease, perpetuating chronic microglial-induced inflammation of the CNS and exacerbating tau pathology [196]. Aβ and NFT can lead to activation of Toll-like receptors and the NRLP3 inflammasome, inducing microglia to generate pro-inflammatory mediators [196]. Systemic inflammation can induce aberrant LPA signaling, which is known to cause microglia polarization and, consequently, neuroinflammation through MAPK-dependent pathways [197]. In addition, LPA can promote BV-2 and primary murine microglia to convert into a pro-inflammatory M1-like phenotype [143]. In mice with experimental autoimmune encephalomyelitis, increased ATX and LPA levels were found in plasma and spinal cord. Furthermore, ATX was found to be expressed in CD11b+ cells (microglia and macrophages), and genetic deletion of ATX from these cells improved the progression of the disease [198].
5. Future Directions: ATX/LPA Axis as a Potential Therapeutic Target
ATX inhibition has enormous potential, including possible applications in pathologies like brain cancer, pruritus, pain-related dysfunctions, or neurodegenerative diseases [199], but we are still beginning to explore this field and further research is needed. Two oral ATX inhibitors, GLPG1690 and FTP-180, are currently on trial for idiopathic pulmonary fibrosis [200,201] and ziritaxestat, another orally administered ATX inhibitor, for systemic sclerosis [202]. On the other hand, a phase three clinical trial for ziritaxestat has failed to improve outcomes in patients with idiopathic pulmonary fibrosis [203].
New ATX inhibitors have been designed recently, of which the non-zinc-binding ATX inhibitor BIO-32546 is selective, orally bioavailable, and capable of penetrating into the brain, and has demonstrated in vivo efficacy for acute pain [204]. More recently, cannabinoids have been used as ATX inhibitors, the most potent of them being MEY-003, which has been found to inhibit in vitro (but it has potential to be orally bioavailable) both ATX β and ATX γ, making it a possible therapeutic agent to treat neurological disorders [205]. Another ATX inhibitor derived from ursodeoxycholic acid has also been recently developed, which targets the hydrophobic tunnel and active site of the phosphodiesterase domain [206]. Fingolimod, a well-known oral drug used to treat multiple sclerosis, whose main mechanism of action is the modulation of the S1P receptors, has been found to inhibit ATX [207,208]. In mice, this has resulted in peripheral nerve regeneration after sciatic nerve crush, particularly in wildtype C57BL/6 and Foxn1 (-/-), but not in Rag1 (-/-) [209]. Furthermore, the ATX inhibitor PF-8380, with potential oral bioavailability, improved neuroinflammation in mouse models of hepatic encephalopathy, reducing levels of LPA 18:0 in the cerebral cortex, hippocampus, brain edema, and plasma, consequently improving motor and cognitive functions [210]. Currently, no ATX inhibitor has been tested specifically for AD.
The actions of LPA1 have also been targeted for therapeutic strategies. The effects of gintonin, a glycolipoprotein fraction of ginseng that is enriched in LPA [211], are also mediated by LPA1. Gintonin has been the focus of recent research because it is a natural, easy-to-obtain product with several potential therapeutic applications, there being strong evidence of its neuroprotective character [212]. Its administration seems safe and might improve the condition of cognitively impaired elderly people [213]. Gintonin could allegedly promote non-amyloidogenic processing to restore brain function when administered orally in mice with AD (AβPPswe/PSEN-1 double Tg mice) [214]. Gintonin-mediated LPA can also activate the LPA1/BDNF/TrkB/Akt pathway to attenuate oxidative damage caused by iodoacetic acid [215], a possible risk factor of AD. Its therapeutic action could also be explained through its possible role in neuronal morphological changes and migration by activating the LPA1/3 receptors [216]. Moreover, it can increase BBB permeability, improving the delivery of drugs into the CNS [217], one of the largest obstacles for the treatment of neurological diseases. Specifically, the intravenous coadministration of gintonin with donepezil, a cognition-improving drug used in AD [218], enhances donepezil brain delivery through gintonin action over the LPA1/3 and VEGF receptors [219].
There are also monoclonal antibodies against LPA in the early phases of preclinical studies, such as Lpathomab and its derivative, called 504B3, which shows potential for improving patient outcomes following TBI when given intravenously in C57BL/6 J mice [220,221]. B3 was also tested both in vitro and in vivo, by subcutaneous administration in zebrafish, to analyze its capacity to improve spinal cord injury outcomes. It has been shown to effectively block LPA1–3 receptors, to decrease inflammation in scar tissue, and to promote neuronal cell survival and synaptic density [222].
As of today, no clinical trials have been initiated using ATX/LPA axis inhibitors for the treatment of AD.
6. Final Remarks
This review highlights the pivotal role of the ATX/LPA axis signaling pathway in the pathophysiology of AD. This signaling cascade, through both direct and indirect mechanisms, emerges as a crucial player in several pathological processes, particularly contributing to neurodegeneration.
The components comprising the ATX/LPA axis exhibit promising potential not only as contributors to our understanding of AD but as essential elements for potential diagnostic biomarkers and therapeutic targets. Early diagnosis is crucial for implementing effective treatment strategies and potentially slowing down the degenerative processes in the central nervous system.
Further research focused on the roles of ATX and LPA in neurological diseases, particularly AD, is imperative. Deeper insights into the intricate interactions within the central nervous system will not only expand our understanding of the pathophysiology of AD but open avenues for the development of targeted therapeutic approaches.
In conclusion, the ATX/LPA axis emerges as a key signaling cascade in the complex landscape of AD. Its multifaceted involvement in pathological processes positions it as a promising area for future research, offering potential breakthroughs in both diagnostics and therapeutics, ultimately aiming to enhance our ability to tackle this challenging neurodegenerative disorder.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alzheimer’s Association 2021 Alzheimer’s Disease Facts and Figures Alzheimer’s Dement.20211732740610.1002/alz.1232833756057 · doi ↗ · pubmed ↗
- 2Mc Dade E.M. Alzheimer Disease Contin. Lifelong Learn. Neurol.20222864867510.1212/CON.000000000000113135678397 · doi ↗ · pubmed ↗
- 3Gallardo G. Holtzman D.M. Amyloid-β and Tau at the Crossroads of Alzheimer’s Disease Adv. Exp. Med. Biol.2019118418720310.1007/978-981-32-9358-8_1632096039 · doi ↗ · pubmed ↗
- 4Custodia A. Ouro A. Romaus-Sanjurjo D. Pías-Peleteiro J.M. de Vries H.E. Castillo J. Sobrino T. Endothelial Progenitor Cells and Vascular Alterations in Alzheimer’s Disease Front. Aging Neurosci.20221381121010.3389/fnagi.2021.81121035153724 PMC 8825416 · doi ↗ · pubmed ↗
- 5Heneka M.T. Carson M.J. Khoury J.E. Landreth G.E. Brosseron F. Feinstein D.L. Jacobs A.H. Wyss-Coray T. Vitorica J. Ransohoff R.M. Neuroinflammation in Alzheimer’s Disease Lancet Neurol.20151438840510.1016/S 1474-4422(15)70016-525792098 PMC 5909703 · doi ↗ · pubmed ↗
- 6Huang W.J. Zhang X. Chen W.W. Role of Oxidative Stress in Alzheimer’s Disease (Review)Biomed. Rep.2016451952210.3892/br.2016.63027123241 PMC 4840676 · doi ↗ · pubmed ↗
- 7Crivelli S.M. Giovagnoni C. Visseren L. Scheithauer A.L. de Wit N. den Hoedt S. Losen M. Mulder M.T. Walter J. de Vries H.E. Sphingolipids in Alzheimer’s Disease, How Can We Target Them?Adv. Drug Deliv. Rev.202015921423110.1016/j.addr.2019.12.00331911096 · doi ↗ · pubmed ↗
- 8Custodia A. Romaus-Sanjurjo D. Aramburu-Núñez M. Álvarez-Rafael D. Vázquez-Vázquez L. Camino-Castiñeiras J. Leira Y. Pías-Peleteiro J.M. Aldrey J.M. Sobrino T. Ceramide/Sphingosine 1-Phosphate Axis as a Key Target for Diagnosis and Treatment in Alzheimer’s Disease and Other Neurodegenerative Diseases Int. J. Mol. Sci.202223808210.3390/ijms 2315808235897658 PMC 9331765 · doi ↗ · pubmed ↗