Protocol for end-design-free rebooting of terminally redundant Pseudomonas phages in clinical isolates of Pseudomonas aeruginosa
Daigo Yokoyama, Nana Kimura, Haruka Yamamoto, Yoshiaki Sakata, Jumpei Fujiki, Hidetomo Iwano

TL;DR
This paper provides a detailed protocol for rebooting Pseudomonas phages with a specific genome structure using clinical isolates of Pseudomonas aeruginosa.
Contribution
A novel protocol for rebooting Pseudomonas phages with a terminally redundant, circularly permuted genome using clinical isolates.
Findings
Steps for constructing a 65 kbp Pseudomonas phage DNA genome through seamless in vitro assembly.
Phage DNA purification and electroporation-based rebooting using clinical isolates of P. aeruginosa.
Successful synthesis of phages with terminally redundant and circularly permuted genomes.
Abstract
Synthetic phage platforms are robust microbiology tools with therapeutic potential against antimicrobial-resistant bacteria. Here, we present a protocol for rebooting Pseudomonas phages with a terminally redundant, circularly permuted 65 kbp genome. We describe steps for designing PCR primers to generate DNA fragments, reconstituting the complete linear phage genome, performing seamless in vitro assembly, and finally, purifying and electroporating the DNA using a P. aeruginosa clinical isolate. •Steps for constructing 65 kbp Pseudomonas phage DNA by genome assembly•Preparation of competent cells for phage rebooting with P. aeruginosa clinical isolates•Phage DNA purification and electroporation-based rebooting of linear phage genomes•Synthesis of phages with terminally redundant and circularly permuted genomes Steps for constructing 65 kbp Pseudomonas phage DNA by genome assembly…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
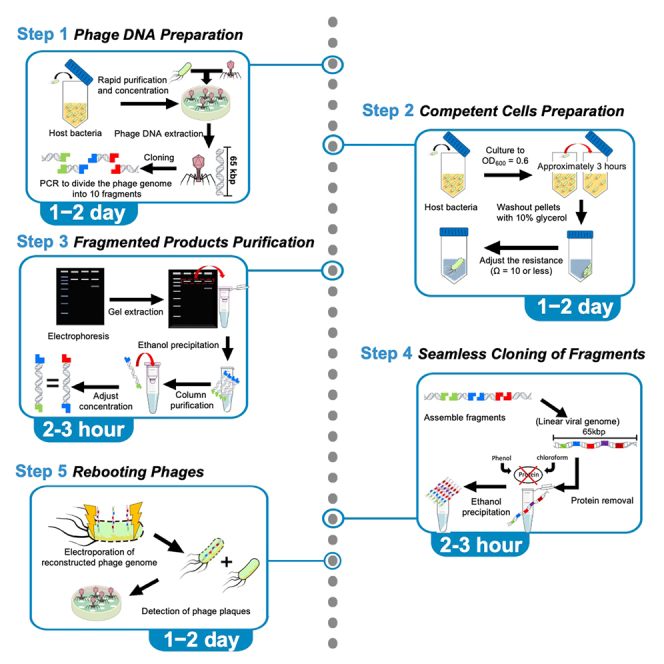 Figure 1
Figure 1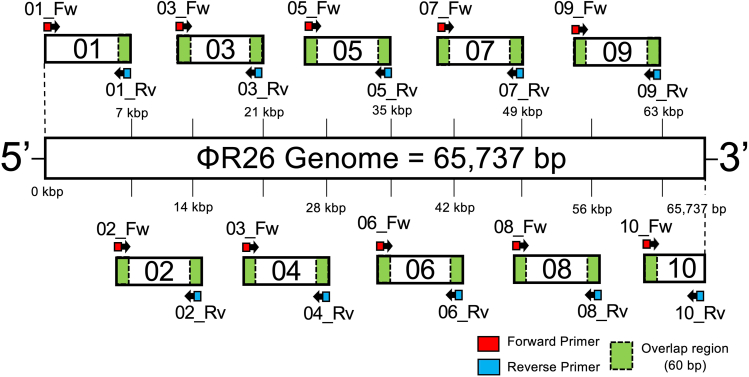 Figure 2
Figure 2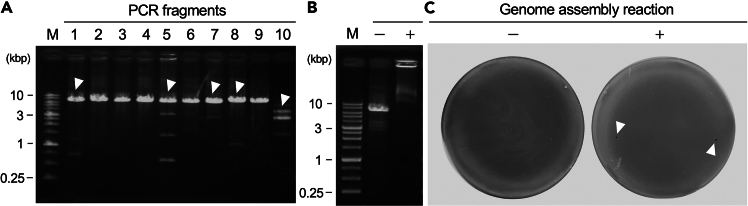 Figure 3
Figure 3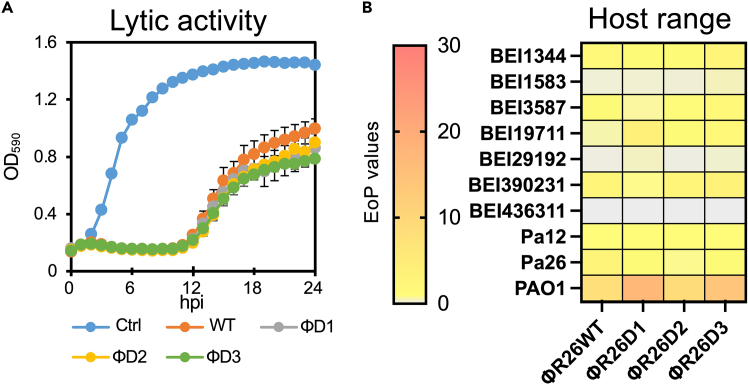 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacteriophages and microbial interactions · Vibrio bacteria research studies · Bacterial biofilms and quorum sensing
Before you begin
In recent years, the widespread use of antibiotics in western countries has overshadowed phage therapy.1 Nevertheless, with the rapid increase in the proportion of antimicrobial-resistant bacteria, phage therapy has once again gained attention as a powerful therapeutic option.2^,^3 Moreover, synthetic phage platforms are providing critical insights that drive innovation and advancement in phage therapy.4^,^5^,^6 However, major challenges in rebooting synthetic phages are genome size and defense systems against phages,7^,^8 because these factors make the process more technically inefficient, less convenient and inhibit phages proliferation. This highlights the need for a straightforward, benchtop protocol for rebooting synthetic phages, given the rise of synthetic phage engineering. In this context, we established an optimized, rapid, and efficient rebooting protocol without cell-free systems9^,^10 or L-form bacteria11^,^12 and a phage defense deficient strains,8 using a seamless cloning approach13 for assembling the complete terminally redundant 65 kbp genome of Pseudomonas phage ΦR26 from 10 DNA fragments.
ΦR26 is a member of the Pbunavirus genus which has been estimated as circularly permuted genomes with terminal redundancy.14^,^15^,^16 In phages with this genomic structure, the packaging mechanism does not require fixed genome termini, suggesting that it allow us to design of DNA fragments flexibly—assembly can begin from virtually any point in the genome without disrupting functional elements. This property significantly simplifies genome reconstruction compared to phages with direct terminal repeats (DTR) or cohesive ends, where fragment design must preserve exact end structures. Consistent with these reports, we also estimated the termini of ΦR26 using PhageTermVirome17 and confirmed that it exhibits terminal redundancy (Figure S1). Importantly, the complete genome of ΦR26 was assembled and successfully rebooted using linear DNA fragments without requiring any special design of viral genomic termini. Our results demonstrate that terminally redundant, circularly permuted phages like ΦR26 are particularly suited for seamless assembly and rebooting, offering a robust platform for synthetic phage applications.
Innovation
Synthetic phage engineering requires robust protocols for genome reconstruction. This protocol introduces a major innovation by enabling end-design-free assembly and benchtop rebooting of synthetic Pseudomonas phages without relying on phage-defense-deficient hosts,8 cell-free systems,9^,^10 or L-form bacteria.11^,^12 Unlike phages with defined termini such as DTRs or cohesive ends, circularly permuted phages (e.g., Pseudomonas virus ΦR26) allow for flexible genome fragmentation and seamless assembly, as any internal site can serve as a junction without disrupting essential genes. This flexibility simplifies genome reconstruction and enables efficient rebooting using a clinical P. aeruginosa isolate, thereby expanding the applicability of phage rebooting and accelerating synthetic phage biology and phage therapy.
Institutional permissions
Experiments using synthetic bacteriophages and Pseudomonas aeruginosa strains were conducted under Biosafety Level 2 (BSL-2) conditions in the Rakuno Gakuen University BSL-2 facility in accordance with the Rakuno Gakuen University Biosafety Guidelines.
Key resources table
REAGENT or RESOURCESOURCEIDENTIFIERBacterial and virus strainsP. aeruginosa Pa12Laboratory Resource Library3^,^18GenBank: NZ_CP136598.1Pseudomonas phage R26 (ΦR26)Laboratory Resource Library19GenBank: NC_048663Chemicals, peptides, and recombinant proteinsProteinase KTakara BioCat#9034MgSO_4_·7H_2_ONacalai tesqueCat#:21003-75NaClNacalai tesqueCat#31320-05GelatinSigma-AldrichCat#G1890Tris-HCl pH 7.4Nippon GeneCat#313-90415TryptoneNacalai tesqueCat#32756-85Yeast extractNacalai tesqueCat#15838-45Agarose SNippon GeneCat#312-01193BD Bacto AgarBecton, DickinsonCat#63-6531-35SeaKem ME AgaroseLonzaCat#50011PrimeSTAR MaxTakara BioCat#R045ANovel Juice Supplied in 6× Loading BufferGeneDireXCat#LD001-1000One Mark DNA electrophoresis markerGeneDireXCat#DMF13-01001-kb DNA ladder RTUGeneDireXCat#SD010-R500EtachinmateNippon GeneCat#312-0179199.5% EthanolWako Pure ChemicalCat#057-00451Phenol:Chloroform:Isoamyl alcohol 25:24:1 mixed, pH 7.9Nacalai tesqueCat#25970Chloroform:Isoamyl alcohol 24:1Sigma-AldrichCat#C0549Ethidium bromide (EtBr) solutionNippon GeneCat#315-900511 M Tris-HCl (pH 8.0)Nippon GeneCat#314-90065QuantiFluor dsDNA SystemPromegaCat#E2670Critical commercial assaysQIAquick PCR Purification KitQIAGENCat#28106QIAquick Gel Extraction KitQIAGENCat#28704Phage DNA Isolation KitNorgenBiotekCat#46800TURBO DNA-free KitThermo Fisher ScientificCat#AM1907OligonucleotidesR26_01_Fw 5′-TTCCCTGGCCGCAACATC-3′This paperN/AR26_01_Rv 5′-CGGTGCTTCCACGCC G-3′This paperN/AR26_02_Fw 5′-TGGACTCACGACGCAATTGC-3′This paperN/AR26_02_Rv 5′-TGGAGCTGTTGCCGAGCG-3′This paperN/AR26_03_Fw 5′-TCGGCATCGATACCGACAAC-3′This paperN/AR26_03_Rv 5′-CAGCTCGTCGAGAAAGACG-3′This paperN/AR26_04_Fw 5′-CCGCAGTTTGTCGAGATTTC-3′This paperN/AR26_04_Rv 5′-GAAAATTTCTCTGATCAGTTACACC-3′This paperN/AR26_05_Fw 5′-GGGTTGATTTAGTGCCCAGG-3′This paperN/AR26_05_Rv 5′-AGCATACTCGTCCTGCGC-3′This paperN/AR26_06_Fw 5′-ATCATGACCAGCCTCGACG-3′This paperN/AR26_06_Rv 5′-CGGTCAGTCCGACCGATC-3′This paperN/AR26_07_Fw 5′-CATGTTCAGATGGACCGTTTC-3′This paperN/AR26_07_Rv 5′-GGCCTGACGGAAAGGTTC-3′This paperN/AR26_08_Fw 5′-TCCAAAATGCAGTTCCGGAGAA-3′This paperN/AR26_08_Rv 5′-ATCGGACAGGTTCGTATCGA-3′This paperN/AR26_09_Fw 5′-GGCGTGAAAGTGATTGGCAT-3′This paperN/AR26_09_Rv 5′-CTAGTGATGAAACCGCCTTG-3′This paperN/AR26_10_Fw 5′-TCAGGGCGCGGCCGG T-3′This paperN/AR26_10_Rv 5′-CTGATAGTACATGTAGTTCTGCG-3′This paperN/AOtherPetri dish with vents, 94 × 16 mm, sterileGreinerCat#633181Tube, 50 mL, PP, Screw CapGreinerCat#227261QSP Microtube 1.5 mL clear 39.5 mmThermo Fisher ScientificCat#62-7023-67DISMIC 25CS045AS 39122140ADVANTECCAT#63-1237-36Centrifugal filter unit (15 mL/100000NMWL)Merck MilliporeCat#2-7112-29Sapphire filter pipette tips, 200 μLGreinerCat#775363Filter pipette tips 10 μLNIPPON GeneticsCat#FGF-10SLAFilter pipette tips 1,000 μLNIPPON GeneticsCat#FGF-100020.2 mL 8 strips PCR tube and DomeCapNIPPON GeneticsCat#FG-028DCNEPA cuvette electrode set 2 mm gap 40∼350 μLNEPA geneCat#EC-002SDisposable bacteria spreaderKenisCat#1-318-0059Gel maker set-HR [heat-resistant type]Takara BioCat#GM-HRBioSpec-nanoShimadzuCat#206−26300−31Micro-cooled centrifugeKubotaCat#3700High-speed, large-capacity cooling centrifugeKubotaCat#7000TOYOBO Fas-III UV transilluminatorToyoboCat#9372959C1000 Thermal CyclerBio-RadCat#1851138BLooK LED Trans illuminatorGeneDireXCat#BK001Quantus FluorometerPromegaCat#E6150
Materials and equipment
Preparation of media and reagents
Sodium magnesium (SM) buffer: The following reagents are weighed, and ddH_2_O is added to dissolve the reagents, followed by autoclaving to sterilize (121°C for 20 min)ReagentFinal concentrationVolumeMgSO_4_·7H_2_O2.0 g/L2.0 gNaCl5.8 g/L5.8 gGelatin0.1 g/L0.1 g1 M Tris-HCl pH 8.050 mM50 mLddH_2_ON/A950 mLTotal1,000 mLStore at 20°C–25°C for up to 1 year. Luria–Bertani (LB) broth: The following reagents are weighed, and ddH_2_O is added to dissolve the reagents, followed by autoclaving to sterilize (121°C for 20 min)ReagentFinal concentrationVolumeTryptone10 g/L10 gNaCl10 g/L10 gYeast extract5 g/L5 gddH_2_ON/A1,000 mLTotal1,000 mLStore at 20°C–25°C for up to 2 months. 50× Tris-acetate-EDTA (TAE) bufferReagentFinal concentrationVolumeTris base2 M242 gAcetic acid1 M57.1 mLEDTA disodium salt dihydrate50 mM18.6 gddH_2_ON/A1,000 mLTotal1,000 mLStore at 20°C–25°C for up to 1 year. LB top agar: The following reagents are weighed, and ddH_2_O is added to dissolve the reagents, followed by autoclaving to sterilize (121°C for 20 min)ReagentFinal concentrationVolumeTryptone10 g/L10 gNaCl10 g/L10 gYeast extract5 g/L5 gAgarose ME5 g/L5 gddH_2_ON/A1,000 mLTotal1,000 mLNote: Store at 50°C for up to 2 months. LB agar plate: The following reagents are weighed, and ddH_2_O is added to dissolve the reagents, followed by autoclaving to sterilize (121°C for 20 min)ReagentFinal concentrationVolumeTryptone10 g/L10 gNaCl10 g/L10 gYeast extract5 g/L5 gAgar15 g/L15 gddH_2_ON/A1,000 mLTotal1,000 mLNote: Store at 4°C for up to 2 months. Note: Pour the prepared solution into a flat Petri dish and allow it to stand at room temperature until it solidifies. Note: If water droplets are present on the flat culture medium after cold storage, dry it thoroughly before use.
Step-by-step method details
Phage stock preparation
Timing: 2 days
The purpose of this section is to prepare a phage stock by rapid purification for the extraction of phage DNA in the following sections.
- 1.Incubate host bacteria overnight in LB broth at 37°C.
- 2.Dilute the phage stock to the desired dilution rate in SM buffer. Note: Diluted phages (approximately 10^6^ PFU/mL) can be used.
- 3.Warm LB agar (3 plates) in an incubator.
- 4.Prepare 3 mL of LB top agar in each tube and keep warm at 50°C.
- 5.Mix 110 μL of host bacteria cultured overnight and 110 μL of phage diluent in a 1.5-mL Eppendorf tube.
- 6.Add 200 μL of mixture (prepared in Step 5) to 3 mL of LB top agar placed at 50°C.
- 7.As a negative control, mix 100 μL of host bacteria with 3 mL of LB top agar.
- 8.Before the prepared LB top agar solidifies, quickly pour it over the LB agar and tilt the plate to distribute it evenly. Let the plate sit undisturbed until the agar solidifies completely.
- 9.Once the agar has solidified, invert the plate and incubate it overnight at 37°C. Note: The solidification time is approximately 5 min, and if the agar is not fully solidified, the LB top agar may shift or detach from the plate surface.
- 10.The next day, check the LB agar plates and pick up those that are lysed in a lacy manner.
- 11.Add 3 mL of SM buffer to each of the three LB agar plates showing lace-like lysis and incubate them at 37°C for at least 30 min.
- 12.Collect the lysate solution from the LB agar medium into a 50-mL centrifuge tube by gently scraping it off the top agar using a disposable bacteria spreader.
- 13.Centrifuge the top agar mixed with the lysate solution at 6,000 × g for 10 min.
- 14.Filter-sterilize the supernatant lysate solution using a 0.45-μm syringe filter unit (Advantec, Tokyo, Japan).
- 15.Carefully pour the supernatant lysate into a centrifugal filter unit (Merck Millipore, Burlington, MA) and add 10 mL of SM buffer.
- 16.Securely close the lid and place the centrifuge tube in the centrifuge with the filter face oriented outward, and centrifuge it at 4,000 × g for 10 min.
- 17.Check the filtration level of the lysate solution and centrifuge until 1 mL remains in the upper chamber. Repeat the centrifugation briefly, ensuring that the lysate solution does not completely drain into the lower layer.
- 18.Repeat the same procedure three times, substituting with SM buffer. Note: Ensure that the lysate in the upper chamber does not completely drain into the lower chamber.Note: Because the filtration speed varies based on the type and titer of the phage, it is important to frequently monitor the progression of filtration by performing short centrifugation intervals.
- 19.When only 1 mL of lysate remains in the upper chamber, discard the lower layer and carefully collect 1 mL of the phage solution in the upper chamber into a 1.5-mL Eppendorf tube simultaneously washing the inner filter section with the remaining phage solution in the upper chamber.
- 20.Store the collected phage solution at 4°C. Note: The collected phage solution will maintain titers for approximately 1 month, depending on the type of phage.Note: Phage samples stored for more than 1 month may have decreased titers; hence, it is recommended to check the titers again.
Plaque assay for phage titer determination
Timing: 2 days
This section aims to determine the current titer of the phage sample. In the rebooting of ΦR26, accurate evaluation of the phage titer was essential for obtaining a sufficient concentration of phage DNA using an appropriate method. Although this protocol was optimized for ΦR26, the approach can be effectively applied to other phages as well, making it a fundamental step in the phage rebooting process.
- 21.Incubate host bacteria overnight at 37°C in LB broth.
- 22.Dilute the phage stock to the desired dilution rate in SM buffer. Note: If you have no idea of the phage titer, it is recommended to prepare 10 dilutions ranging from 10^2^ to 10^10^.
- 23.Warm three sheets of LB agar plates at 37°C.
- 24.Heat autoclaved LB top agar in a heat block at 50°C to prevent gelation.
- 25.Mix 110 μL of host bacteria cultured overnight and 110 μL of phage diluent in a 1.5-mL Eppendorf tube.
- 26.Add a 200 μL of mixture (prepared in Step 25) to 3 mL of LB Top Agar.
- 27.As a negative control, mix 100 μL of host bacteria with 3 mL of LB top agar.
- 28.Before the prepared LB top agar solidifies, quickly pour it over the LB agar and tilt the plate to distribute it evenly. Let the plate sit undisturbed until the agar solidifies completely.
- 29.Once the agar has solidified (approximately 5 min), invert the plate and incubate it overnight at 37°C.
- 30.Count the phage plaques and measure the phage titer represented as plaque-forming units per milliliter (PFU/mL). Note: Bubbles formed when LB top agar is layered over LB agar may be mistaken for plaques. To avoid this, mark or remove them by bursting them with a micropipette tip or other suitable method.
Extraction of phage DNA
Timing: 6 h
This section describes the method for extracting the phage genome from the obtained phage stock. In the rebooting of ΦR26, obtaining high-purity, mutation-free DNA at a sufficient yield was crucial for successful genome reconstruction. Although this protocol was optimized for ΦR26, it can also be applied to other phages. Ensuring the integrity and purity of the extracted DNA is a fundamental requirement for phage rebooting, making this step a critical component of the overall process.
- 31.Prepare 900 μL of sample adjusted to 10^10^ PFU/ml of the phage solution obtained in the previous section. Use SM buffer for phage dilution.
- 32.Digest exogenous DNA such as bacterial genomes in the sample using the TURBO DNA-free kit (Thermo Fisher Scientific) according to the manufacturer’s protocol.
- 33.Add 20 μL of Turbo DNase and 100 μL of DNase buffer to the sample and incubate it at 37°C for at least 30 min.
- 34.Add 4 μL of DNase inactivation buffer and incubate the sample at room temperature with 2–3 times of inverted mixing occasionally for 5 min.
- 35.Centrifuge the sample at 10,000 × g for 2 min and collect the supernatant except for the precipitated DNase inactivation buffer.
- 36.Extract the phage DNA using the phage DNA isolation kit (Norgen, Thorold, ON, Canada) according to the manufacturer’s protocol.
- 37.Determine the DNA concentration of the phage genome sample using Biospec-Nano (Shimadzu, Kyoto, Japan).
- 38.Dilute the phage genome sample with nuclease-free water to 100 ng/μL.
- 39.Store the phage genome sample at −20°C. Pause point: For long-term storage, we recommend storing the sample at −80°C.Note: Repeated freezing and thawing can damage the phage DNA. To minimize this risk, store the samples in small aliquots to reduce the frequency of freezing and thawing.
Designing DNA fragments
Timing: 1–2 h
This section describes the design of DNA fragments for seamless cloning. Specifically, we describe the approach of dividing the genome into 10 fragments for PCR amplification, as applied to the Pseudomonas virus ΦR26. Although this method was optimized for ΦR26, the same design principles can be applied to other phages, enabling seamless genome assembly across different phages.Note: If the target phage is registered in GenBank or a similar database, its sequence can be obtained and used from the National Center for Biotechnology Information database.Note: In phages with terminal redundancy, it is possible to freely design the starting point for genome assembly. However, care must be taken to avoid disrupting the integrity of coding sequences (CDSs). Fragment boundaries should be placed outside of annotated CDS regions to ensure functional gene preservation.
- 40.Divide the entire phage sequence into some fragments to be designed (ends design-free), and set up an overlap region of 60 bp.
- 41.To ensure efficiency in the subsequent seamless cloning, adjust the length per fragment to approximately 7 kbp.
- 42.Design primer pairs for amplifying each fragment of the phage genome (Figure 1).Figure 1. Primer design for amplifying PCR fragments from the ΦR26 genomePrimers for ΦR26 genome were designed to provide a 60-bp overlap (green blocks) between each fragment. For the ΦR26 genome, which consists of 65,737 bp, primers were designed to divide the entire genome into 10 fragments. Fragments 1–9 have an approximate product size of 7 kbp, whereas fragment 10 is designed to be 2,737 bp. Arrows with red blocks indicate the forward primers used to amplify each fragment, and arrows with blue blocks indicate reverse primers.
- 43.For ΦR26 of 65,737 bp, there should be 9 fragments of approximately 7 kbp and 1 fragment of 2,737 bp. Note: In this case, only the final fragment (fragment 10) will be short; however, this is not a problem because the concentration can be adjusted during seamless cloning.Note: No overlapping sequences are required on the 5′ side of fragment 1 and 3′ side of fragment 10.Note: Each primer should be approximately 20 bases long, and the Tm values of the primer sets that amplify the same fragment should be set as close to the same level as possible.Note: One successful genome assembly was initiated at the CDS for a structural protein (Figure 1), while an alternative construct beginning with the CDS for a non-structural protein (DNA polymerase) was also successfully rebooted in our additional validation.
Cloning and purification of phage DNA fragments
Timing: 1 day
This section describes the cloning and purification of phage DNA fragments. In the rebooting of ΦR26, obtaining highly purified phage genome fragments with sufficient yield was the most critical factor. Therefore, this step played a vital role in the successful reconstruction of ΦR26 genome. Although this protocol was optimized for ΦR26, it can be broadly applied to other phages as well. Considering its central importance in ensuring the quality and efficiency of phage genome assembly, this section serves as a vital component in the overall phage rebooting process.
- 44.Set up the PCR system as follows: PCR reaction master mixReagentAmountTemplate DNA (10 ng/μL)1–2 μLPrime Star Max Ver.225 μLForward primer (2 μM)3 μLReverse primer (2 μM)3 μLddH_2_OUp to 50 μLTotal50 μLPCR cycling conditionsStepsTemperatureTimeCyclesInitial Denaturation98°C120 s1 CycleDenaturation98°C10 s35 CycleAnnealing65°C45 sExtension72°C120 sFinal extension72°C120 s1 CycleHold4°CForeverNote: As the required annealing temperature varies depending on the designed primers, it is strongly recommended to precisely determine the optimal temperature using gradient PCR.Note: If the yield of DNA is insufficient, it may be improved by extending the extension reaction time.
- 45.Mix 1 g of agarose S (Nippon Gene, Tokyo, Japan) with TAE buffer. Heat the solution to completely dissolve agarose S in the TAE buffer, and prepare an agarose gel.
- 46.Pour TAE buffer with agarose S into the electrophoresis bath so that the top of the agarose gel is soaked approximately 1 mm.
- 47.Mix 10 μL of the DNA fragment sample with 2 μL of 6× loading buffer (Takara Bio, Shiga, Japan).
- 48.Apply 1-kb DNA ladder RTU (GeneDireX, Taoyuan, Taiwan) to the wells of the gel.
- 49.Set the electrophoresis conditions to 100 V for 35 min. Note: If the temperature of the electrophoresis bath exceeds 40°C, the electrophoresis image may become unclear. To prevent this, replace the TAE buffer in the bath with fresh buffer if the bath overheats during the process.Note: If the gel used for electrophoresis is too thick, the electrophoresis image may become unstable, and the background may become overly bright after ethidium bromide staining, thereby making it difficult to observe the bands.
- 50.After the completion of electrophoresis, carefully remove the gel and immerse it in ethidium bromide solution.
- 51.Stain the gel in ethidium bromide solution for 15 min. CRITICAL: Because EtBr is photosensitive, cover the container with aluminum foil.
- 52.Irradiate the stained DNA with UV light and check the image (Figure 2A).Figure 2. Seamless cloning and rebooting of the ΦR26 genome(A) Electrophoretic analysis of ΦR26 DNA fragments amplified by PCR. Fragments 1–9 exhibited lengths of approximately 7,000 bp each, whereas fragment 10 resulted in approximately 2,700 bp as expected. White arrow heads indicate the bands that should be extracted.(B) Electrophoretic profile of the genome assembly product.(C) After electroporation, the assembled genome was introduced into the P. aeruginosa Pa12 strain by electroporation, and phage rebooting was confirmed via a plaque assay. White arrow heads indicate rebooted ΦR26 plaques produced on the lawn of the Pa12 strain.
- 53.Check the electrophoresis image and perform gel extraction and purification only if nonspecific bands are observed. Note: Subsequent experiments can be performed even if nonspecific bands are present. However, it may affect the efficiency of the desired assembly during seamless cloning. Conversely, gel extraction may reduce the sample yield.
For gel extraction (if required).
- 54.Mix Novel Juice 6× loading buffer (GeneDireX) with the remaining 40 μL of the DNA fragment sample not soaked in EtBr for gel extraction.
- 55.Mix 1 g of SeaKem ME Agarose (Lonza, Basel, Switzerland) with TAE buffer. Heat the solution to completely dissolve ME agarose in TAE buffer.
- 56.After mixing the Seakem ME Agarose (Lonza), prepare the gels for electrophoresis using a gel maker (Mupid, Tokyo, Japan) so that the thickness is 3–5 mm.
- 57.Pour 1× TAE buffer into the electrophoresis tank so that the top of the gel is soaked approximately 1 mm.
- 58.Apply the DNA fragment sample mixed with Novel Juice to the electrophoresis wells.
- 59.Set the electrophoresis conditions to 100 V for 35 min.
- 60.Cut out the desired bands using a scalpel under a BLooK LED Transilluminator. Note: To prevent DNA damage, we recommend using fluorescence-based imaging during gel extraction.
- 61.Extract DNA from the gel according to the protocol of the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany).
- 62.Purify all DNA fragment samples according to the protocol of the QIAquick PCR Purification Kit (Qiagen).
- 63.Elute the purified DNA fragments in 100 μL of nuclease-free water.
- 64.Add 3.3 μL of 3 M sodium acetate and 2 μL of etachinmate (Nippon Gene) to each reaction solution.
- 65.Mix the sample by vortexing.
- 66.Add 250 μL of 100% EtOH and mix thoroughly by vortexing.
- 67.Centrifuge the sample at 12,000 × g for 5 min and discard the supernatant. Note: If the desired concentration cannot be achieved from a single gel extraction, perform multiple extractions and concentrate the DNA using ethanol precipitation. To maximize assembly efficiency, ensure that the molar variation between fragments is within ±0.005 pmol.Note: At this time point, do not aspirate the precipitated white pellet DNA by mistake.
- 68.Add 1 mL of 70% EtOH and pipette gently to peel off the DNA precipitate adhering to the wall surface.
- 69.Centrifuge the sample at 12,000 × g for 2 min and discard the supernatant.
- 70.Turn the tube upside down and air-dry for 15 min.
- 71.When sufficiently evaporated, dissolve the DNA in 30 μL of nuclease-free water.
- 72.Measure the DNA concentration using Biospec-Nano (Shimadzu).
- 73.Dilute the DNA with nuclease-free water to a concentration of 0.05 pmol/fragment. Note: The DNA purification step in this section serves to eliminate the template phage DNA used during PCR, thereby preventing unintended rebooting of the original template phage. Our validation confirmed that the sequential purification procedures effectively reduced residual template DNA to undetectable levels.
Seamless cloning and purification of the assembled product
Timing: 1–2 days
We successfully reconstructed the complete genome (65 kbp) of Pseudomonas virus ΦR26 from the 10 purified PCR-amplified DNA fragments. In this section, we describe an efficient seamless cloning method for assembling DNA fragments and detecting cloned DNA segments. Using this approach, the complete genomes of other phages can also be reconstructed. The long DNA molecules generated in this process must be highly accurate and yield high efficiency. Therefore, this study describes a method for the precise cloning of DNA fragments and obtaining a complete phage genome with high yield.
- 74.Mix the DNA fragment sample prepared in the “cloning and purification of phage DNA fragments” section in the following volume (for 10 fragments): DNA assembly reaction systemReagentFinal concentrationAmountDNA fragment0.05 pmol1 μL/fragmentNEBuilder HiFi DNA Assembly Master Mix1×10 μLNuclease-free waterN/AMix up to 10 μL if the DNA fragments are <10 μLTotalN/A20 μLCRITICAL: The DNA fragments used for the seamless cloning of 10 fragments should be adjusted to high purity to improve the efficiency of assembly. Ensure to check for the absence of protein or ethanol contamination using a spectrophotometer or similar instrument. The recommended purity is OD260/280 = 1.75–1.9 and OD260/230 ≥2. If these conditions are not met, it is recommended to repeat the column purification in Step 62.CRITICAL: The DNA fragments used for the seamless cloning of 10 fragments can be adjusted to equimolar concentration to improve the yield of the desired fragments. Ideally, the molar concentration of each fragment should be adjusted to within ±0.005 pmol using a spectrophotometer.
- 75.Incubate the sample mixture in a thermal cycler at 50°C for 2 h. Note: Although the general protocol requires 1 h at 50°C for the assembly of DNA fragments, we recommend extending the reaction time to 2 h to increase the yield.
- 76.Mix agarose S to 0.8% in TAE buffer. The solution is heated to completely dissolve the ME agarose in 1× TAE buffer.
- 77.After mixing agarose S, prepare the gels for electrophoresis using a gel maker (Mupid) so that the thickness is 3–5 mm.
- 78.Pour 1× TAE buffer into the electrophoresis bath so that the top of the gel is soaked approximately 1 mm.
- 79.Mix 1 μL of 6× loading buffer and 5 μL of the assembly product.
- 80.Apply 1-kb DNA Ladder RTU (GeneDireX) and the assembled product stained with 6× loading buffer to the wells of the agarose gel.
- 81.Set electrophoresis conditions to 100 V for 45 min.
- 82.After electrophoresis, stain with EtBr for 15 min for imaging, and check the integrity of DNA fragment ligation (Figure 2B).
- 83.To purify the assembled phage DNA for rebooting, mix up 15 μL of the assembly product not used for imaging to 100 μL with nuclease-free water.
- 84.Add equal volumes of phenol:chloroform:isoamyl alcohol (PCI) (Nacalai Tesque, Kyoto, Japan).
- 85.Pipette gently with a micropipette.
- 86.Centrifuge at 18,000 × g for 5 min at room temperature, and discard the supernatant. Note: Be careful not to aspirate the lower layer.
- 87.Add an equal volume of chloroform:isoamyl alcohol (CIA) (Nacalai Tesque) to the collected supernatant.
- 88.Pipette gently with a micropipette.
- 89.Centrifuge at room temperature, 18,000 × g for 5 min, carefully remove the aqueous layer (upper layer), and transfer it to another tube.
- 90.Confirm the purity of the obtained DNA samples using the BioSpec-Nano (Shimadzu) by measuring the OD260/280 and OD260/230 values.
- 91.Accurately quantify the DNA concentration using the Quantus Fluorometer (Promega, Madison, WI) with the QuantiFluor dsDNA System (Promega), according to the manufacturer’s protocol. CRITICAL: If proteins are not removed by PCI and CIA treatment, they may cause issues such as arcing during electroporation.
Preparation of competent cells from P. aeruginosa clinical isolates
Timing: 1 day
This section describes a rapid and reproducible protocol for preparing competent cells from the P. aeruginosa clinical isolate Pa12, optimized for electroporation of linear DNA fragments.
- 92.Inoculate 5 mL of LB broth with 20 μL of Pa12 glycerol stock and incubate overnight at 37°C with shaking.
- 93.Mix with 100 μL of Pa12 incubated overnight with 9.9 mL of LB broth (100-fold dilution).
- 94.Incubate the sample at 37°C for 3–4 h with shaking until the OD_600_ reaches 0.6.
- 95.Centrifuge the sample at 10,000 × g for 5 min and remove the supernatant, except for the pellet, by aspiration.
- 96.Pipette the pellet slowly in sterile water and resuspend.
- 97.Refill 1 mL of the suspension into another container.
- 98.Centrifuge it at 10,000 × g for 5 min, remove the supernatant, except for the pellet, by aspiration, and resuspend it by pipetting slowly with 1 mL of 10% glycerol solution. Perform this process twice. Note: To obtain a competent cell with the appropriate resistance, ions must be separated from the sample into the supernatant. It is recommended to pipette approximately 100 times with a 1-mL pipette tip to separate the ions.Note: If the measured resistance is too high, adding a small amount of TE buffer (Tris-EDTA) can help reduce the resistance.
- 99.Centrifuge the sample at 10,000 × g for 5 min, remove the supernatant, except for the pellet, by aspiration, add 500 μL of 10% glycerol solution gently over the pellet, transfer 100 μL to a cuvette, and measure the resistance value. Note: We recommend checking the actual resistance values of the competent cells. The recommended resistance is <10 kΩ. If the sample resistance is too high or too low, the appropriate voltage may not be applied to the competent cell.Note: Sucrose-based preparation of competent P. aeruginosa cells has also been reported to yield high electroporation efficiency,20 and we also successfully used this method to reboot phages in Pa12. However, our protocol is specifically optimized for the delivery of linear DNA fragments, which are more prone to intracellular degradation. To ensure sufficient DNA uptake per cell, a moderate cell density is required. Therefore, competent cells obtained from overnight cultures with high biomass may be suboptimal for this application.Note: Because the freeze–thaw cycle reduces transformation efficiency, it is recommended that the prepared competent cells be divided into smaller aliquots.
Storage: Competent cells can be stored for approximately 6–12 months at −80°C with high electroporation efficiency.
Gene transfer by electroporation
Timing: 2 days
In this section, we describe how to introduce a complete phage genome toward the competent cell obtained in the previous section.
- 100.Inoculate 5 mL of LB broth with 20 μL of host bacteria and incubate it overnight at 37°C with shaking.
- 101.Measure 100 μL of the overnight-incubated host bacteria, mix it with 9.9 mL of LB broth, and dilute the culture 100-fold.
- 102.Incubate the diluted culture at 37°C with shaking for 3–4 h until the OD_600_ reaches 0.6.
- 103.Retrieve the competent cells prepared in the previous section from the stock and thaw them on ice.
- 104.Gently invert the thawed competent cells to mix them.
- 105.Add 100 μL of the competent cells to a NEPA cuvette electrode set and measure the resistance.
- 106.Add 200 ng of the complete reconstructed phage genome to the competent cells in the cuvette.
- 107.Set up a negative control by adding the same amount of solvent for the reconstructed phage genome.
- 108.Measure the resistance and confirm that it falls within the range of <10 kΩ. Note: Check for a slight decrease in resistance after the inserted DNA has been mixed with the competent cell sample.Note: If the resistance is >10 kΩ, add 1 μL of TE buffer to the sample, check the resistance each time, and adjust it until the appropriate value is reached.
- 109.Incubate the entire cuvette in ice for 5 min.
- 110.Perform electroporation under the following conditions: Electroporation systemVoltage (V)Pulse width (ms)Pulse interval (ms)Number of times2,6003.5501Note: If necessary, a recovery culture of electroporated cells can be performed for approximately 3 h in SOC medium.
Mix 100 μL of the electroporated sample in the NEPA cuvette with 100 μL of the nonelectroporated Pa12 cells prepared in Step 102), and plate the mixture as described. In parallel, mix 100 μL of the same DNA sample with 100 μL of the competent Pa12 cells without electroporation as a negative control to check for possible phage contamination. No plaques should be observed in this control if the DNA preparation is free of phage particles.CRITICAL: In this protocol, untreated bacterial cells are mixed with electroporated bacterial cells. The untreated bacteria are responsible for compensating for the dead bacteria in electroporation and forming a lawn that facilitates the identification of phage plaques.
- 111.Prepare two autoclaved LB agar plates.
- 112.Place the LB top agar in a 50°C heat block to prevent solidification.
- 113.In a 1.5-mL tube, combine the mixed sample prepared in Step 110), add 200 μL of the sample to 3 mL of LB top agar, and mix the sample thoroughly.
- 114.Before the LB top agar solidifies, quickly overlay it onto the LB agar plate, tilting the plate to ensure an even distribution. Allow the agar to set until it completely solidifies.
- 115.Once the agar has solidified (approximately 5 min), invert the plate and incubate it overnight at 37°C.
- 116.Inoculate 5 mL of LB broth with 20 μL of host bacteria and incubate it overnight at 37°C with shaking.
- 117.The following day, examine the plate for plaque formation.
- 118.If plaque formation is observed or the entire surface is lysed, it indicates that the phage has been successfully rebootted (Figure 2B). Note: If plaque is observed on the negative control, it is recommended to reconsider the entire rebooting process, as plaque formation may be due to phage contamination or possible prophage induction caused by electroporation stress.
Cloning and rapid propagation of rebooted phages
Timing: 3 days
This section describes how to purify and use the phage successfully rebooted in the previous section.
- 119.If plaques are identified, prepare pipette tips for picking by cutting off the tip of the pipette tip and autoclaving it at 121°C for 20 min.
- 120.Collect the plaque with the pipette tip for picking with the entire surrounding Pa12 lawn, place it in a tube containing 1 mL of LB broth, and incubate it overnight with shaking.
- 121.Centrifuge the sample at 10,000 × g for 5 mins, and collect 800 μL of the coculture supernatant in a 1.5-mL tube.
- 122.Filter-sterilize 800 μL of the coculture supernatant with a sterile 0.45-μm syringe filter and collect the phage solution.
- 123.Repeat the same procedure three times to ensure that they are clonal isolates.
- 124.In order to amplify clonal isolates, rebooted phages will be propagate and purified as described in the “phage stock preparation” section.
Expected outcomes
This protocol is designed to enable rebooting Pseudomonas virus ΦR26 with DNA of 65 kbp. Several additional experiments should be performed to confirm that the parent and rebooted phages are exactly similar. Here is an example of exactly how to check if the phage rebooted by electroporation is completely identical to the parent phage. After whole-genome sequencing with 150-base paired-end reads of three rebooted clones of ΦR26 using DNBSEQ-G400 (MGI Tech, Shenzhen, China), using extracted phage DNA (as described in the “extraction of phage DNA” section), we evaluated the completeness of the phage genome compared with parental ΦR26. No single nucleotide variants, insertions, or deletions were detected (data not shown). The use of high-fidelity polymerase is critical to ensure sequence accuracy sufficient for reliable phage rebooting. Moreover, as depicted in Figure 3A, rebooted ΦR26 resulted in almost complete similar lytic activity against the propagating strain Pa12 compared with parental ΦR26 in the turbidity reduction assay as described elsewhere19 Furthermore, host range determination based on the efficacy of the plating (EoP) method21^,^22 showed that the killing spectrum of rebooted ΦR26 was completely similar to that of parental ΦR26 against representative 10 strains of P. aeruginosa (Figure 3B). The representative plaque-forming activity of parental ΦR26 and the rebooted phage (ΦR26D1) is illustrated in Figure S2. These examples demonstrate that our protocol can accurately reboot ΦR26 from its DNA reconstructed by the assembly of 10 PCR fragments in vitro. This protocol could be applied to generate phages with specific modifications by incorporating any desired genetic sequence during the fragment design stage in the future. Moreover, by examining the sequences of lysogenic phages in pathogenic bacteria and designing fragments to remove the repressor region,23 it is possible to convert lysogenic phages into virulent phages.Figure 3. Validation of the lytic activity of rebooted ΦR26 strains(A) Lytic curves of parental ΦR26 and rebooted three ΦR26 strains, assigned as ΦD1, ΦD2, and ΦD3, respectively. Phages were inoculated into the Pa12 strain at an MOI of 0.1, and OD_590_ values were monitored every 1 h for 24 h at 37°C with shaking using a plate reader (Sunrise Rainbow Thermos RC, TECAN, Grödig Austria). The individual points in each lytic curve are presented as means ± SD (n = 3).(B) Phage host range determination performed using the EoP method. P. aeruginosa strains (Pa12 and Pa26 derived from animal infection sites,3^,^18^,^19 PAO1 purchased from ATCC, BEI strains derived from P. aeruginosa BEI panel24) were grown in LB medium overnight at 37°C with shaking. LB top agar containing P. aeruginosa strains was overlaid on an LB agar plate. Thereafter, 2 μL of diluted parental or rebooted ΦR26 aliquot (10^7^–10^1^ PFU/mL) in SM buffer was dropped onto an overlaid LB agar plate to observe the lytic activity of phages by plaque formation. EoP values represent PFU using the specific P. aeruginosa strain/PFU using Pa12.
Limitations
This protocol summarizes the workflow for rebooting Pseudomonas phages with genomes of approximately 65 kbp. Nevertheless, the conditions for each step must be adjusted depending on the host bacterium and phage strain that you will use. Phages and host strains are highly diverse, and their properties can vary significantly. Therefore, the experimental conditions should be flexible and adaptable. Another potential limitation of this protocol is the loss of DNA due to repeated purification steps aimed at improving the purity of DNA fragments and the viral genome simultaneously minimizing sample concentration errors as much as possible through close monitoring and essential optimization of DNA extraction. Typically, 2–3 plaques are detected on the lawn of the host P. aeruginosa strain per agar plate following electroporation. However, in some instances, no plaques may form. Therefore, preparing multiple plates for each rebooting experiment is recommended. Furthermore, plaque detection efficiency can be improved by using a host strain in which key phage defense systems, such as restriction-modification, CRISPR systems and CBASS, have been deleted or attenuated as described by Ipoutcha et al. for rebooting of Pbunaviruses.8
Troubleshooting
Problem 1
Insufficient amount of phage genomic DNA is acquired.
Potential solution
- •One of the simplest solutions is to strengthen the titer of the phage. Simply increasing the phage titer will improve the yield of phage genomic DNA.
- •If you collect the eluate from the elution tube and pass it through the column again, you may be able to recover any residual DNA on the column.
Problem 2
Electrophoresis image is unclear.
Potential solution
- •Check that the gel thickness is appropriate. If the gel is too thick, staining may brighten the background and make it difficult to observe the bands.
- •If the DNA amount is too small, increase the concentration by ethanol precipitation to increase the concentration.
- •Check to ensure that the electrophoresis bath is not excessively heated. Overheating of the electrophoresis bath may cause a temperature difference between the surface and center of the gel, which may result in unclear images.
- •Check that the staining time with ethidium bromide is appropriate. If the staining time is too long, the background may become brighter, making it difficult to identify the bands. If the staining time is too short, the DNA emission may be weak, and it may be difficult to identify the band.
Problem 3
Resistance of the competent cell is low or the addition of DNA causes a large decrease in resistance.
Potential solution
- •The major cause of low resistance of the competent cell is insufficient desalination. Gentle pipetting during competent cell preparation is important to sufficiently dissociate salts adhering to the cells into the solution and remove them as supernatant. If the resistance of the competent cell is low, increase the frequency of pipetting or ensure that the pellet is sufficiently resuspended in the solution.
- •If the resistance decreases significantly with the addition of DNA, the DNA may not have been sufficiently purified. Ensure that impurities have been adequately removed through column purification or phenol/chloroform treatment. Furthermore, DNA purification using dialysis membranes, in addition to this protocol, may help maintain appropriate resistance values.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jumpei Fujiki ([email protected]).
Technical contact
Technical inquiries regarding this protocol should be directed to and will be answered by the technical contact, Jumpei Fujiki ([email protected]).
Materials availability
Materials associated with this protocol are available upon request from the lead contact.
Data and code availability
This protocol does not report datasets or generate code.
Acknowledgments
The authors are grateful to Dr. Tomohiro Nakamura (Azabu University, Japan, and University of California San Diego, US) for his suggestions and helpful advice. This study was supported by grants for Scientific Research on Innovative Areas and International Group from the MEXT/10.13039/501100001691JSPS KAKENHI (JP23K23791).
Author contributions
Conceptualization, D.Y. and J.F.; data curation, D.Y., J.F., N.K., H.Y., and H.I.; formal analysis, D.Y. and J.F.; funding acquisition, J.F. and H.I.; investigation, D.Y., N.K., H.Y., and J.F.; methodology, Y.S. and J.F.; project administration, J.F.; resources, Y.S., J.F., and H.I.; software, J.F.; supervision, J.F. and H.I.; validation, N.K., H.Y., Y.S., J.F., and H.I.; visualization, D.Y. and J.F.; writing – original draft, D.Y. and J.F.; writing – review and editing, N.K., H.Y., Y.S., J.F., and H.I.
Declaration of interests
The authors declare no competing interests.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Summers W.C.The strange history of phage therapy Bacteriophage 2201213013310.4161/bact.2075723050223 PMC 3442826 · doi ↗ · pubmed ↗
- 2Fujiki J.Nakamura K.Nakamura T.Iwano H.Fitness trade-offs between phage and antibiotic sensitivity in phage-resistant variants: molecular action and insights into clinical applications for phage therapy Int. J. Mol. Sci.2420231562810.3390/ijms 242115628 PMC 1065065737958612 · doi ↗ · pubmed ↗
- 3Nakamura K.Fujiki J.Nakamura T.Furusawa T.Gondaira S.Usui M.Higuchi H.Tamura Y.Iwano H.Fluctuating bacteriophage-induced gal U deficiency region is involved in trade-off effects on phage and fluoroquinolone sensitivity in Pseudomonas aeruginosa Virus Res.306202119859610.1016/j.virusres.2021.19859634648885 · doi ↗ · pubmed ↗
- 4Kiga K.Tan X.E.Ibarra-Chávez R.Watanabe S.Aiba Y.Sato'o Y.Li F.Y.Sasahara T.Cui B.Kawauchi M.Development of CRISPR-Cas 13a-based antimicrobials capable of sequence-specific killing of target bacteria Nat. Commun.112020293410.1038/s 41467-020-16731-632523110 PMC 7287087 · doi ↗ · pubmed ↗
- 5Lu T.K.Collins J.J.Dispersing biofilms with engineered enzymatic bacteriophage Proc. Natl. Acad. Sci. USA 1042007111971120210.1073/pnas.070462410417592147 PMC 1899193 · doi ↗ · pubmed ↗
- 6Mitsunaka S.Yamazaki K.Pramono A.K.Ikeuchi M.Kitao T.Ohara N.Kubori T.Nagai H.Ando H.Synthetic engineering and biological containment of bacteriophages Proc. Natl. Acad. Sci. USA 1192022 e 220673911910.1073/pnas.2206739119 PMC 986024836409909 · doi ↗ · pubmed ↗
- 7Cheng L.Deng Z.Tao H.Song W.Xing B.Liu W.Kong L.Yuan S.Ma Y.Wu Y.Harnessing stepping-stone hosts to engineer, select, and reboot synthetic bacteriophages in one pot Cell Rep. Methods 2202210021710.1016/j.crmeth.2022.100217 PMC 914268935637913 · doi ↗ · pubmed ↗
- 8Ipoutcha T.Racharaks R.Huttelmaier S.Wilson C.J.Ozer E.A.Hartmann E.M.A synthetic biology approach to assemble and reboot clinically relevant Pseudomonas aeruginosa tailed phages.Microbiol. Spectr.122024 e 028972310.1128/spectrum.02897-23PMC 1091338738294230 · doi ↗ · pubmed ↗