Efficient iterative CRISPR/Cas9 editing using sid-1 co-conversion and feeding RNAi in Caenorhabditis elegans
Alexandra S Weisman, Nicole M Fisher, Craig P Hunter

TL;DR
This paper introduces a method to efficiently create complex genetic strains in C. elegans using CRISPR/Cas9 and sid-1 RNAi techniques.
Contribution
A novel CRISPR/Cas9 co-conversion strategy using sid-1 RNAi for iterative gene editing in C. elegans.
Findings
Using sid-1 loss-of-function CRISPR reagents reduces candidate screening from thousands to tens of F1 progeny.
Restoration-of-function CRISPR reagents enable visible RNAi phenotype screening for efficient strain development.
The optimized sid-1 CRISPR design increases the success rate of co-conversion events for multiple gene edits.
Abstract
We present a sid-1 loss-of-function and restoration-of-function CRISPR/Cas9 co-conversion protocol in Caenorhabditis elegans. Introducing CRISPR reagents that induce sid-1 loss-of-function can produce survivors on lethal RNAi foods while reagents that induce sid-1 restoration-of-function can be screened for restoration of visible RNAi phenotypes. Both methods efficiently reduce the pool of candidates from hundreds or thousands of F1 progeny to tens with minimal experimenter effort. Furthermore, our optimized sid-1 CRISPR design allows a high ratio of CRISPR reagents targeting the gene of interest, maximizing successful co-conversion events. The interconvertibility of the sid-1 locus readily enables this strategy to be leveraged to iteratively create complex strains with multiple gene edits.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
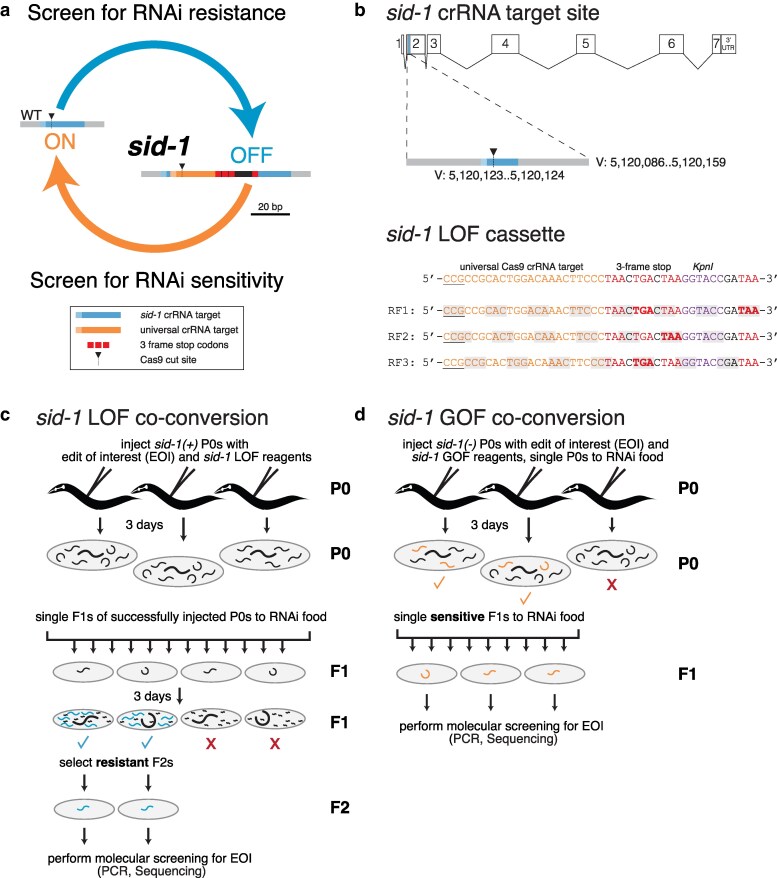 Fig. 1
Fig. 1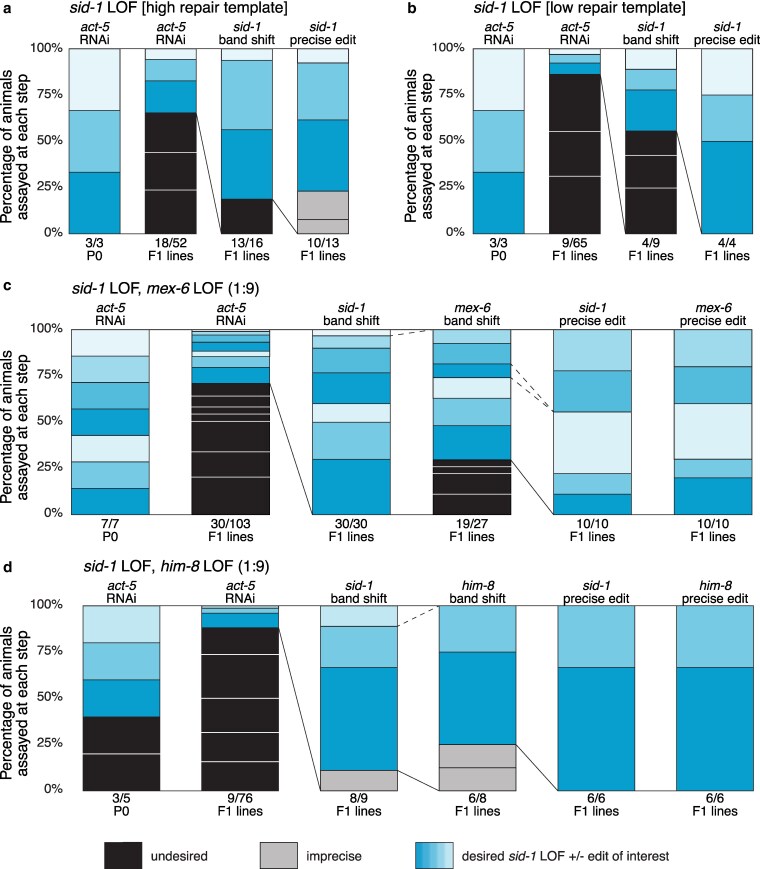 Fig. 2
Fig. 2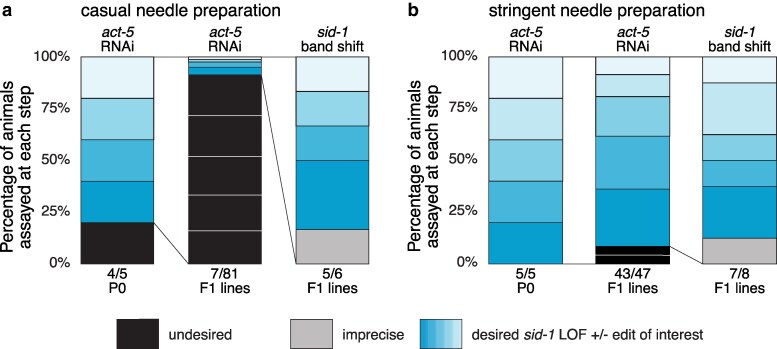 Fig. 3
Fig. 3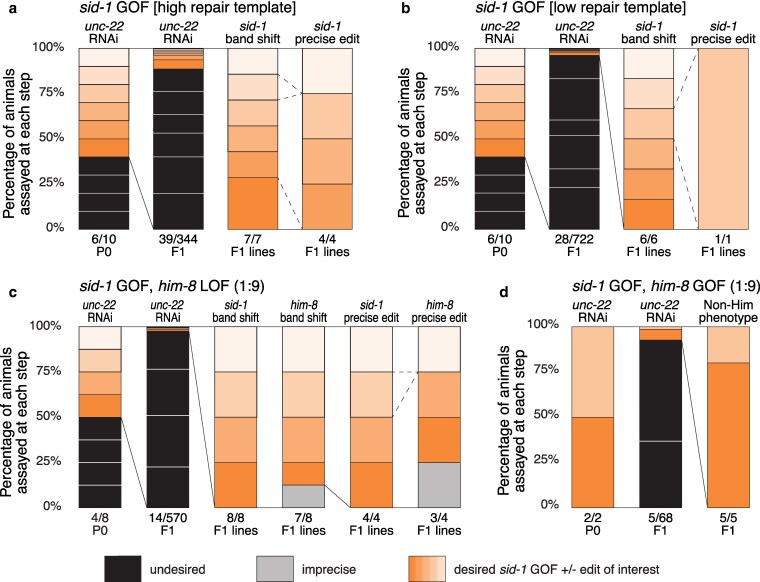 Fig. 4
Fig. 4- —National Institutes of Health10.13039/100000002
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCRISPR and Genetic Engineering · Genetics, Aging, and Longevity in Model Organisms · Evolution and Genetic Dynamics
Introduction
Co-CRISPR and co-conversion are widely used in Caenorhabditis elegans genome editing to confirm reagent functionality and identify animals likely to contain successful edits of interest. However, many of these strategies introduce mutations whose phenotypes complicate downstream analyses and thus must be removed prior to subsequent experiments (Arribere et al. 2014; Kim et al. 2014). Rescue of temperature-sensitive pha-1(e2123) restores the wild-type sequence but is only compatible with strains that can tolerate 25°C growth (Ward 2015). A recent publication employs an iterative, or “flip-flop”, Dumpy/non-Dumpy selection procedure, yet one must then inject Dumpy animals (Lins et al. 2023). These challenges can make co-CRISPR/conversion schemes inefficient for iterative or complex genome-editing workflows.
We have developed highly efficient sid-1 Cas9 cleavage and repair reagents that make sid-1, when combined with feeding RNAi, a highly effective co-conversion marker. Feeding RNAi is a widely used technique, and the food-dependent phenotype can be tailored to experimenter needs and experience. Loss-of-function (LOF) mutations in sid-1 confer RNAi resistance, providing a clear, selectable phenotype, while restoration-of-function (ROF) sid-1 alleles restore RNAi sensitivity, providing numerous choices for visible phenotypes (Winston et al. 2002). These phenotypes can be assessed rapidly in the F1 and F2 progeny, providing immediate feedback on the preparation and injection of the CRISPR reagents. If sid-1 co-conversion succeeds but a co-injected edit of interest (EOI) fails, the issue is likely due to guide RNA or repair template design rather than reagent handling. Additionally, identifying candidates with precise edits among hundreds of offspring is time-consuming and labor-intensive. Our findings demonstrate that the vast majority of animals with sid-1 co-conversions also harbor the desired edit in the gene of interest, significantly reducing the number of progeny that must be screened. Finally, the sid-1 locus can be readily and rapidly restored to wild-type function, eliminating the co-CRISPR phenotype and/or enabling the introduction of additional EOIs. This reiterative cycling between sid-1 LOF and sid-1 ROF enables generating complex genome edits. By combining simplicity, reliability, and versatility, sid-1 co-conversion offers a transformative approach for genome editing in C. elegans, facilitating both single and sequential edits with minimal technical barriers.
Materials and methods
C. elegans maintenance
Strains were maintained at 20°C on normal growth medium plates seeded with OP50 (Brenner 1974).
Design and optimization considerations of CRISPR guides and repair templates
One (or 2) CRISPR RNA (crRNA) and a single-stranded oligodeoxynucleotide (ssODN) repair template containing two ∼35 nucleotide (nt) homology arms were used for LOF and ROF cassettes. When possible, target sequences with the recommended NG nucleotides upstream of the protospacer adjacent motif (PAM) were selected for higher editing efficiency (Farboud et al. 2019). See Supplementary file for exact sequences of crRNAs and ssODN repair templates.
sid-1 LOF alleles (qt160 and qt164, see Supplementary file) were generated by a crRNA (sid-1 AG) that targets early in the wild-type sequence and a 117 nt anti-sense ssODN repair template. Design elements from the Meyer, Seydoux, and Sternberg groups were combined to maximize the theoretical efficiency of the sid-1 LOF repair (Paix et al. 2016, 2017; Wang et al. 2018; Farboud et al. 2019). Both sid-1 LOF homology arms are proximal/terminal, immediately abutting the double-stranded DNA break (Paix et al. 2016). As in Wang et al. (2018), the repair template directs insertion of a universal cassette containing an exogenous Cas9 target sequence, stop codons in all 3 frames, and a restriction site, as well as inducing a downstream frameshift. Our modifications include the following: (1) switching the NheI site to KpnI to permit enzyme activity directly in Phusion PCR buffer, (2) further optimizing the inserted exogenous crRNA target sequence from Paix et al. (2017) (UcrRNA_AW1) to increase the off-target score without affecting the on-target score (Integrated DNA Technologies 2019, RRID:SCR_025813), (3) changing the 2 nucleotides upstream of the PAM from AG to GG to improve ribonucleoprotein (RNP) cutting efficiency, and (4) inverting the orientation of the exogenous crRNA target sequence within the cassette to improve the repair efficiency of subsequent sid-1 ROF edits: inversion relative to the Wang et al. (2018) STOP-IN cassette consolidates the recessed homology to one arm (6 nt and 39 nt recession instead of 17 nt and 28 nt).
To convert sid-1 LOF to sid-1 ROF (wild-type), a crRNA targeting the inserted exogenous target sequence (UcrRNA_AW1) and a 73 nt ssODN consisting entirely of homology arms were used.
The mex-6 deletion used 2 crRNAs targeted near the start codon (mex-6 ATG) and within the 3′ UTR (mex-6 UTR) (Sternberg et al. 2024). A 107 nt ssODN repair template directed insertion of a stop cassette and our exogenous crRNA (UcrRNA_AW1) target sequence to produce a precise 2219 nt deletion.
him-8 was targeted for insertion of a reversible LOF cassette, similar to the sid-1 LOF cassette, by injecting a single crRNA (him-8 AA) and a 108 nt ssODN repair template (Sternberg et al. 2024). To convert him-8 LOF to him-8 ROF (wild-type), a crRNA targeting the inserted exogenous target sequence (UcrRNA_AW1) and a 67 nt ssODN consisting entirely of homology arms were used.
CRISPR reagent preparation
We generally followed the Star Protocol from the Mello group and recommend using that as a detailed reference (Ghanta et al. 2021). Our modifications to their protocol include: (1) ssODN repair template concentration (1 µL of 40 pmol/μL stock was added), (2) following assembly of the full injection mix, we centrifuge at 13,000*×g* for 2 minutes to avoid needle clogging, transfer 19 μL to a fresh tube and keep on ice, (3) 0.5 μL of mixture was loaded into an injection needle and injections were performed until at least 3 high-quality injections (of both gonad arms) have been achieved. After centrifugation, sid-1:EOI reagents were combined at a 1:9 ratio. Reagent mixtures were stored at 4°C. Animals were recovered on a common plate for 6 hours before transferring individual P0 s to fresh plates.
Stringent needle hygiene
CRISPR RNP injection needle hygiene begins with stringent chain of custody of capillaries: new capillaries (freshly opened from manufacturer) were aliquoted in small batches into 15 mL polypropylene screw cap tubes while wearing gloves that were treated with RNase-away (or similar product). A similarly treated brand-new razor blade was used to cut a hole in the cap just large enough for a single capillary to slide through. Aliquoted capillaries were stored in an RNase-free area.
Prior to pulling injection needles, a Kimwipe wetted with RNase-away was used to swab all portions of the needle puller that may contact the capillary. After 1 minute of contact time, the same portions of the puller were rinsed with a Kimwipe wetted with RNase-free water, then dried. We similarly treated the needle holder on the injection rig. Since our puller and rig are also used for non-RNase-free injections, we repeated this procedure each time prior to pulling new needles.
Care was taken to only handle capillaries and pulled needles with RNase-away treated gloves, and only in the middle; we did not touch the ends. A fresh capillary was shimmied through the hole in the cap of the tube, loaded into the needle puller, and the program was run. The freshly pulled needles were rested on clay supports in an RNase-away treated petri dish and humidified by a Kimwipe saturated with RNase-free water. Good RNase-free technique and tips were used to load needles with injection mix (we backloaded quick-fill filamented capillaries, 1B100F-4, World Precision Instruments (RRID:SCR_008593)). Loaded needles were fitted into the needle holder of the injection rig with RNase-away treated gloves. From this point on standard (casual) injection techniques were used (Halocarbon/paraffin oil, buffers), after all, worms are not RNase-free.
Injection considerations to facilitate troubleshooting
When possible, we followed F1 offspring from P0 bearing F1 roller frequencies above 5% and moderate fertility (∼80–200 offspring over 48 hours) but note that we also recovered co-CRISPR and edits of interest from animals with no F1 rollers or low fertility (as few as 18 offspring). Since the sid-1 ROF approach facilitates screening of the whole F1 brood it obviates identification of the cohort within the brood that was exposed to the injection mix (rollers). However, users may still wish to include the pRF4::rol-6 plasmid to distinguish issues with microinjection technique from issues with reagent preparation (Mello et al. 1991).
RNAi food and plate preparation, storage and use
RNAi foods with post-embryonic RNAi phenotypes were selected to avoid confounding results from maternal effects. Clones for *act-5/*Y25C8.2, *unc-22/*ZK617.1 were sourced from the Ahringer Library (Kamath and Ahringer 2003; RRID:SCR_017064). RNAi food was prepared as in Protocol 1 (Kamath et al. 2001) except with 100 µg/mL carbenicillin (Carb) and given 24-hour drying and induction time on isopropyl-β-D-thiogalactopyranoside (IPTG)/Carb plates. Note that IPTG/Carb plates have a limited 4°C shelf life and should be seeded within 1 month after being poured and should be used within 8 weeks of pouring. Whenever RNAi plates are prepared, immediately test wild-type and sid-1 mutant controls to confirm the RNAi food is properly prepared before proceeding with phenotypic scoring within sid-1 co-conversion workflows. Assays were performed at 20°C.
sid-1 LOF workflow using act-5 RNAi resistance
After injected P0 animals have laid progeny on 2 OP50 plates (the first day post-injection and the second day post-injection), score the plates for the proportion of roller progeny. Select 2–4 P0s with the highest roller proportions and moderate fertility. Move the F1 progeny to act-5 RNAi plates when they are L4s to avoid act-5 effects on fertility. It is advisable to single as many F1s as possible to increase the likelihood of recovering edits of interest. Some experiments in this study utilized pools of 2 to 5 F1s at this step to increase the number of F1s scored while maintaining a practical number of RNAi plates (pooled experiments are indicated in figure legends and Supplementary file), in these cases, only a single F1 line was followed from each plate. After 24 hours, remove the adult to produce a semi-synchronized cohort for facile scoring 2 to 3 days later. Identify act-5 RNAi-resistant animals by comparison to age-matched positive (homozygous sid-1 mutant) and negative (wild-type) controls. Only homozygous sid-1 mutants will grow past the L3 stage; therefore, plates with multiple L4/young adults are likely successful co-conversion lines. Single 2–4 sibling F2s from each act-5 resistant plate to fresh plates; once these F2 animals have laid sufficient progeny, the F2 adults can be used directly for PCR genotyping to identify homozygous sid-1 and EOI.
We note that if only lines with imprecise sid-1 edits and precise EOI edits are found, imprecise sid-1 edits can be easily outcrossed using non-lethal RNAi food. If additional sid-1 co-conversion is being used, first confirm sid-1 LOF as a precise insertion with an intact exogenous crRNA target site and PAM by sequencing to ensure it can be converted to ROF.
sid-1 ROF workflow using unc-22 RNAi sensitivity
Recovered (6 hours) injected P0s were singled onto *unc-*22 RNAi plates and transferred on day 2 to a second *unc-*22 RNAi plate for another day and removed. Candidate RNAi-sensitive (twitching) L4 to young adult animals were identified by comparison to positive (wild-type) and negative (homozygous sid-1 mutant) controls. F1 twitchers were singled to produce a subsequent generation on unc-22 RNAi plates. It is advisable to single as many F1s as possible to increase the likelihood of recovering edits of interest. In our hands, well-made unc-22 RNAi plates do not require addition of levamisole to identify twitchers, but animals can be picked into 3 mM levamisole to facilitate scoring (Lewis et al. 1980). To identify potential homozygous sid-1 and EOI, and avoid loss of non-phenotypic EOI edits, isolate ∼12 twitching sibling F2s from each unc-22 F1 plate. Once F2 animals have laid sufficient progeny, the adult F2 can be used directly for PCR genotyping. Once homozygous ROF animals (100% twitching progeny) are identified, RNAi food is no longer required.
Him phenotype scoring
L4 animals were singled and allowed to lay for 24–48 hours and then removed. Scoring the sex of 25–35 adult self-progeny was sufficient to call animals Him or Non-Him. Twelve to 16 F2s per candidate him-8 LOF F1 line were singled to increase the likelihood of recovering homozygous animals that exhibit the Him phenotype.
Band shift analysis and sequencing
F2 animals were used for band shift analysis and F2 or F3 apparent homozygotes were used for Sanger sequencing. Individual adults were lysed as in Minkina and Hunter (2017), except 15 µL of buffer was used per adult and the 95°C step was extended to 15 minutes. PCR was performed in Phusion HF Buffer (New England Biolabs; RRID:SCR_013517) scaled to 20 µL reactions, 3 µL of the worm lysate as template and a standard 30-cycle 3-step PCR (see Supplementary file for cycling parameters, oligo sequences, and amplicon lengths). The precise edit produced by our sid-1 LOF reagents generates a 44 base pair (bp) band shift compared to wild-type and thus is easily visualized by gel electrophoresis on a 2% Agarose/TBE gel. Upon efficient KpnI digestion, wild-type sid-1 amplicons are unchanged, but sid-1 LOF amplicons are cleaved once. This optional digestion step can provide further insight on the precision of repair and inform selection of candidates for sequencing. The candidates selected for sequencing in this study were analyzed without digestion.
The precise mex-6 deletion produces a 2219 bp band shift compared to wild-type. The precise edit our him-8 LOF reagents produce has a 41 bp band shift compared to wild-type.
Sequencing off-target genes
Animals were lysed as above. Four F2s were collected per candidate F1. Quality scores and continuous read length were used to identify poor quality runs; furthermore, individual base quality scores were examined within a 100 nt window of the putative cut site using a custom R script (see data availability statement): runs with any individual quality score <40 within that window were re-run and the traces were manually reviewed. Results from the highest quality run for each F2 are available in the Supplementary file.
Guidance on naming any alleles generated with our co-CRISPR reagents
If using strain HC1292 to convert sid-1(qt160) LOF to ROF, which is molecularly wild-type, the resulting strain should be named *sid-1(YourLabAlleleDesignation##[qt160]) (Tuli et al. 2018).
Strains, plasmids, crRNAs, ssODN repair templates, and oligos used in this study are listed in the Supplementary file.
Results
sid-1 loss-of-function and restoration-of-function strategy
We modified design elements from previously described methods (Paix et al. 2016, 2017; Wang et al. 2018; Farboud et al. 2019) to create a highly efficient and reversible sid-1 allele (Fig. 1a). See methods section for full details and advice on EOI design. The sid-1 LOF was generated by a crRNA that targets the wild-type sequence and a 117 nt ssODN that templates insertion of an exogenous crRNA target site and 3-frame stop knock-in cassette (Fig. 1b and Supplementary file). To convert sid-1 LOF to sid-1 ROF (wild-type), a crRNA that targets the exogenous sequence and a 73 nt ssODN repair template were used. Once the wild-type sequence has been restored, further co-conversion edits are enabled (Fig. 1a). Our screening pipelines begin with either the LOF or ROF RNAi phenotype, proceeds to a PCR band shift assay to identify programmed insertion/deletions, and concludes with selective sequence confirmation.
The iterable sid-1 co-conversion strategy provides a flexible system that enables sequential editing. a) Overview of iterable co-conversion strategy. b) Genomic location of the endogenous sid-1 crRNA target site and sequence of the loss-of-function cassette; all 3 reading frames (RF) are presented. Workflow of c) sid-1 loss-of-function (LOF) and d) restoration-of-function (ROF) co-conversions.
sid-1 LOF CRISPR is effective and easy to identify
To identify sid-1 LOF animals, the F1 offspring of injected N2s were moved to a post-embryonic lethal RNAi food (act-5) at the L4 stage (Fig. 1c). Only homozygous sid-1 LOF animals will grow to adulthood on act-5 RNAi plates. A single F2 adult was selected from each such plate to establish candidate F1 lines for molecular confirmation. We used PCR to identify F1 lines with the expected sid-1 band shift and then sequenced select candidates to confirm precise insertion of the sid-1 LOF cassette.
We observed that 77% (n = 13) of candidate F1 lines had precise edits at the sid-1 locus, not insertions or deletions (Fig. 2a). If RNAi resistance occurs but precise insertions are not observed, it likely indicates issues with the repair template. Lowering the repair template concentration to simulate co-conversion conditions reduced the number candidates but did not adversely affect the recovery of precisely edited sid-1 alleles (Fig. 2b). All P0 animals produced at least one precise sid-1 edit, making identification of candidate lines easy and quick. One sequence-confirmed sid-1 LOF precise edit was designated as qt160 and curated as strain HC1185; its outcrossed descendant, HC1292, is available at the Caenorhabditis Genetics Center (CGC, RRID:SCR_007341). Our sid-1 crRNA has 2 potential off-target sites in the genes linc-165 and pxd-1, both of which have 4 mismatches within the guide sequence (CRISPOR (RRID:SCR_015935); see Supplementary file). While it is unlikely that sites with this many mismatches would be cut, we sequenced them to be sure (Au et al. 2019; Farboud et al. 2019). Concomitant with our guide RNA design principles, we failed to detect any sequence changes within 100 nt of the putative cut sites of the off-target genes for 8 independent F1s in addition to strain HC1185 (see Supplementary file). These results suggest that when screening for edits of interest free of off-target lesions, 2 or 3 independent candidates should suffice unless one is exceedingly unlucky.
sid-1 LOF strategy readily identifies co-conversion candidates. a–d) Each bar shows the percentage of animals of interest in blue (phenotypic or precisely edited), imprecisely edited in gray, and undesired in black (non-phenotypic or unedited animals). Each box within a bar marks an individual P0 and its progeny of interest across the screening steps. Candidates carried forward are indicated by connecting lines; dotted lines indicate a P0 cohort excluded from further analysis. F1s were singled unless otherwise indicated. Two representative experiments are shown for injection mixes containing only sid-1 LOF reagents at high [4 pmol/µL] a) and low [0.4 pmol/µL] b) ultramer repair template concentration. Two representative sid-1 LOF co-conversion experiments identifying candidates for mex-6 (c) or him-8 (d) LOF edits. The results from 2 replicate mex-6 experiments are presented with P0s grouped by experiment: the top 4 P0s (F1s singled) and bottom 3 P0s (F1s plated in pools of 3).
Validating co-CRISPR of sid-1 LOF and edits of interest
We designed CRISPR reagents to precisely edit mex-6 and him-8, 2 germline-expressed genes whose loss-of-function phenotypes should not interfere with identifying or recovering edits of interest. Both genes were targeted for insertion of LOF cassettes containing a triple stop cassette, exogenous crRNA target site and KpnI restriction site similar to the sid-1 LOF cassette. To increase the likelihood that sid-1 edited animals would also harbor the edit of interest, we employed a reagent ratio of sid-1:EOI of 1:9, a more aggressive ratio then utilized in most other methods (Paix et al. 2015, 2016; Farboud et al. 2019; Yang et al. 2020).
The mex-6 gene is functionally redundant with mex-5, thus it can be deleted without causing a phenotype (Schubert et al. 2000). mex-6 coding sequence was replaced with the LOF cassette using 2 crRNAs and a 107 nt ssODN repair template. Precise mex-6 EOIs were readily recovered using sid-1 LOF co-conversion (Fig. 2c). To assess reproducibility, this experiment was performed twice, with RNP mixtures prepared several months apart; combined data are shown. In both experiments, all injected N2 animals produced F1s that laid act-5 RNAi-resistant F2 progeny. All 30 F1 lines produced sid-1 PCR band shifts indicating successful insertion of the repair template and the absence of in/dels at the sid-1 locus. Nineteen F1 lines also exhibited expected mex-6 band shifts, demonstrating deletion of the entire coding region and efficient repair template insertion at the mex-6 locus. Ten F1 lines selected from 5 different P0s were confirmed to have precise edits at both sid-1 and mex-6. In summary, 7 injected P0 animals produced 30 F1 resistant lines, 19 of which contained the expected mex-6 band shift, and all 10 sequenced F1 lines proved to have precise edits at both sid-1 and mex-6.
The him-8 gene is required for proper X-chromosome segregation; null alleles increase the frequency of males (Hodgkin et al. 1979). The him-8 coding sequence was disrupted by inserting an LOF cassette using a single crRNA and a 108 nt ssODN to direct repair. Precise him-8 EOIs were readily recovered using sid-1 LOF co-conversion (Fig. 2d). Three of 5 injected N2 animals produced 9 F1-resistant lines, 8 of which showed the expected sid-1 band shift. Six of these 8 exhibited the expected him-8 band shifts, and all 6 F1 lines had precise edits at both sid-1 and him-8. One sequence-confirmed isolate was curated as HC1189 (him-8(qt163) IV; sid-1(qt164) V). All lines with precise him-8 LOF edits displayed the Him phenotype.
Our results demonstrate that precise sid-1 edits and EOI occur more frequently than imprecise edits and frequently occur together.
Stringent needle hygiene increases editing efficiency
During reagent testing, we found that needle hygiene was a critical factor that influenced CRISPR efficiency. See methods section for a description of proper needle hygiene. When less stringent needle hygiene procedures were followed, the proportion of act-5 resistant F1 lines was dramatically reduced (Fig. 3a). Stringent needle hygiene, including RNAse-free needle aliquoting, storage, pulling, and loading, substantially increased the proportion of candidate F1 lines (Fig. 3b). Although fewer F1 candidates were identified when using casual needle conditions, editing proficiency (precise band shifts) remained high. Therefore, sid-1 LOF co-conversion enables identification of candidate lines even when reagent preparation is suboptimal.
Needle preparation affects editing efficiency but not proficiency. a, b) Each bar shows the percentage of animals of interest in blue (phenotypic or precisely edited), imprecisely edited in gray, and undesired in black (non-phenotypic or unedited animals). Each box within a bar marks an individual P0 and its progeny of interest across the screening steps. Candidates carried forward are indicated by connecting lines; dotted lines indicate a P0 cohort excluded from further analysis. The relative efficiency of sid-1 LOF conversion attempted with casual needle preparation (a) and stringent needle preparation (b) are shown. In both conditions the repair template concentration was 4 pmol/µL and F1s were plated in pools of 2–5.
sid-1 ROF CRISPR is effective and easy to identify
Since a single wild-type copy of sid-1 restores feeding RNAi sensitivity, after a brief recovery, injected animals were singled to a non-lethal RNAi food (unc-22) and F1 progeny were scored for twitching (see Methods) (Fig. 1d). As observed for sid-1 LOF CRISPR, repair template concentration correlates with the number of recovered candidates but does not affect the ability to recover precise sid-1 ROF alleles (Fig. 4a and 4b). Our sid-1 ROF unc-22 results highlight the power of this co-conversion paradigm; simply by scoring for visible twitching, the number of probable candidates were reduced 10- to 20-fold. Our exogenous crRNA has 2 potential off-target sites in the genes clec-83 and faxc-1, both of which have 4 mismatches within the guide sequence (CRISPOR (RRID:SCR_015935); see Supplementary file). While it is unlikely that sites with this many mismatches would be cut, we sequenced them to be sure (Au et al. 2019; Farboud et al. 2019). We failed to detect any sequence changes within 100 nt of the putative cut sites of the off-target genes for 7 independent F1s, thus 2 or 3 independent candidates should suffice unless one is exceedingly unlucky (see Supplementary file).
sid-1 ROF strategy simplifies identification of co-conversion candidates. a–d) Each bar shows the percentage of animals of interest in orange (phenotypic or precisely edited), imprecisely edited in gray, and undesired in black (non-phenotypic or unedited animals). Each box within a bar marks an individual P0 and its progeny of interest across the screening steps. Candidates carried forward are indicated by connecting lines; dotted lines indicate a P0 cohort excluded from further analysis. Two representative experiments are shown for injection mixes containing only sid-1 ROF reagents at high [4 pmol/µL] a) and low [0.4 pmol/µL] b) ultramer repair template concentration. c) sid-1(qt160) animals were injected with reagents to insert a revertible him-8 LOF cassette. d) him-8(qt163); sid-1(qt164) animals were injected with reagents to revert both loci to wild-type; only phenotypes were scored.
Validating co-conversion of sid-1 ROF and edits of interest
To validate sid-1 ROF co-conversion, we used strain HC1185 (sid-1(qt160)V) and the him-8 LOF reagents established earlier (Fig. 2d). We then used sid-1 ROF to repair him-8, using strain HC1189 (him-8(qt163) IV; sid-1(qt164) V) and reagents that revert the him-8 LOF allele (qt163) to wild-type (him-8 ROF).
Precise him-8 LOF alleles were readily identified and recovered among sid-1 ROF animals (Fig. 4c). Specifically, 4 of 8 injected HC1185 (sid-1 LOF) P0s produced 14 unc-22-sensitive F1s and 12 of these gave rise to unc-22-sensitive F2s. Eight of the confirmed 12 F1 lines produced the expected sid-1 band shifts. Seven of these 8 F1 lines also exhibited the expected him-8 band shifts. Four sequenced F1 lines, selected from 4 different P0s, had precise sid-1 reversion and 3 had precise him-8 LOF insertions. As expected, all lines with precise homozygous him-8 LOF edits displayed the Him phenotype.
To test co-conversion, we injected HC1189 (him-8 precise LOF, sid-1 precise LOF) with our exogenous crRNA that targets both genes and 2 gene specific ssODNs to restore both him-8 and sid-1 to their respective wild-type functions. Two injected HC1189 P0s produced 5 unc-22 sensitive F1s, all of which gave rise to unc-22-sensitive F2s that produced a non-Him phenotype, indicating that him-8 function was restored and the locus was precisely reverted to wild-type (Fig. 4d).
Discussion
The sid-1 co-CRISPR protocol described here offers a streamlined approach for genomic editing in C. elegans. Feeding RNAi is inexpensive, ubiquitous, and easy. RNAi phenotypes are conditional on the provided food, thus altering RNAi sensitivity offers multiple options for screening and selection phenotypes; experimenters can select an RNAi phenotype they are experienced scoring that is compatible with their desired edits of interest. Furthermore, the high efficiency of recovering precise edits at the sid-1 locus, likely enabled by reagent optimization including the aggressive sid-1:EOI concentration ratio of 1:9, is tightly coupled to the recovery of precise edits at the target locus, reducing the number of animals that must be injected, screened, and sequenced. If the sid-1 LOF strategy is used, and sid-1 does not affect phenotypic analysis, the edited strain can be analyzed directly, or sid-1 can be readily reverse-edited to wild-type. Lethal RNAi, such as act-5, further simplifies LOF screening by removing all animals without sid-1 editing. As LOF candidates are tested as F2s, there is no likelihood of mosaicism.
The sid-1 ROF strategy restores the sid-1 wild-type sequence in the F1 progeny, thus RNAi sensitive animals can be identified in the F1 generation. As ROF selects for restoration of function this strongly favors the recovery of precise edits compared to any LOF co-conversion scheme. Furthermore, the dominant phenotype identifies progeny exposed to both functional Cas9 reagents and to active C. elegans homology directed repair processes, which likely contributes to the efficient recovery of target gene EOIs. For ROF co-conversion, non-mosaic animals can be easily identified by testing one subsequent generation for RNAi sensitivity. For all these reasons, we strongly recommend using the strain HC1292 and the efficient sid-1 ROF strategy for single EOI designs. To reduce the number of spontaneous mutations (from normal culturing in the lab) prior to deposition at the CGC, the qt160 allele was outcrossed 6 times to N2. Regardless, prudent adopters may wish to sequence any loci they intend to edit and confirm wild-type behavior in any non-RNAi assays before injecting reagents.
Finally, the ease of iteratively cycling between sid-1 LOF and ROF while creating strains with multiple alleles and low incidence of off-target editing eliminates the need to use multiple co-conversion markers or to remove co-conversion marker(s) by outcrossing. While other schemes have 1 or 2 of these benefits, our scheme simultaneously maximizes recovery of desired edits of interest, minimizes experimenter effort and maintains a degree of flexibility not available in previously described strategies. We encourage researchers interested in adopting this approach to obtain strains from the Caenorhabditis Genetics Center (RRID:SCR_007341) and reach out for additional guidance.
Supplementary Material
jkaf128_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Arribere JA, Bell RT, Fu BX, Artiles KL, Hartman PS, Fire AZ. 2014. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas 9 in Caenorhabditis elegans. Genetics. 198(3):837–846. doi:10.1534/genetics.114.169730.25161212 PMC 4224173 · doi ↗ · pubmed ↗
- 2Au V, Li-Leger E, Raymant G, Flibotte S, Chen G, Martin K, Fernando L, Doell C, Rosell FI, Wang S, et al 2019. CRISPR/Cas 9 methodology for the generation of knockout deletions in Caenorhabditis elegans. G 3 (Bethesda). 9(1):135–144. doi:10.1534/g 3.118.200778.30420468 PMC 6325907 · doi ↗ · pubmed ↗
- 3Brenner S . 1974. The genetics of Caenorhabditis elegans. Genetics. 77(1):71–94. doi:10.1093/genetics/77.1.71.4366476 PMC 1213120 · doi ↗ · pubmed ↗
- 4Farboud B, Severson AF, Meyer BJ. 2019. Strategies for efficient genome editing using CRISPR-Cas 9. Genetics. 211(2):431–457. doi:10.1534/genetics.118.301775.30504364 PMC 6366907 · doi ↗ · pubmed ↗
- 5Ghanta KS, Ishidate T, Mello CC. 2021. Microinjection for precision genome editing in Caenorhabditis elegans. STAR Protoc. 2(3):100748. doi:10.1016/j.xpro.2021.100748.34505086 PMC 8417391 · doi ↗ · pubmed ↗
- 6Hodgkin J, Horvitz HR, Brenner S. 1979. Nondisjunction mutants of the nematode Caenorhabditis elegans. Genetics. 91(1):67–94. doi:10.1093/genetics/91.1.67.17248881 PMC 1213932 · doi ↗ · pubmed ↗
- 7Integrated DNA Technologies . 2019. CRISPR design tool [Internet]. Coralville (IA): Integrated DNA Technologies; [accessed 2019 May 30]. Available from https://www.idtdna.com/site/order/designtool/index/CRISPR_SEQUENCE.
- 8Kamath RS, Ahringer J. 2003. Genome-wide RN Ai screening in Caenorhabditis elegans. Methods. 30(4):313–321. doi:10.1016/S 1046-2023(03)00050-1.12828945 · doi ↗ · pubmed ↗