Characterizing fatty acid oxidation genes in Drosophila
Juliana Geronazzo, Abigail Heimerl, Linnea Lindell, Skye McCrimmon, Clara Stormer, Brooke Horvai, Ian P Johnson, Tia M Peterson, Jocelyn Zuckerman, Anna I Scott, Meredith M Course

TL;DR
This study uses fruit flies to investigate genes involved in fatty acid oxidation, helping to understand rare genetic diseases in humans.
Contribution
The study identifies specific Drosophila genes as functional orthologs of human fatty acid oxidation disorder genes.
Findings
Arc42, but not CG4860, mirrors the acylcarnitine profile of human ACADS loss of function.
Mcad is confirmed as the likely ACADM ortholog, and a codon deletion in Mtpα causes deleterious effects.
Loss of function in Etf-QO and CG7834 is homozygous lethal in Drosophila.
Abstract
In this study, we leverage the power and tractability of Drosophila genetics to better understand the molecular mechanisms underlying a group of rare genetic diseases known as fatty acid oxidation disorders. We use CRISPR-Cas9 to generate mutations in 6 putative fatty acid oxidation genes in Drosophila, then analyze the phenotypes and acylcarnitine profiles of these flies. We find that while Arc42 and CG4860 are both predicted orthologs of human ACADS, only Arc42 loss of function mirrors the acylcarnitine profile of ACADS loss of function. Acylcarnitine profiles also support our previous identification of Mcad as the likely ACADM ortholog, and reveal the deleterious effects of a single codon deletion in Mtpα (the predicted human HADHA ortholog). Finally, we observe that loss of function in Etf-QO and in CG7834—predicted orthologs of human ETFDH and ETFB, respectively—is homozygous…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
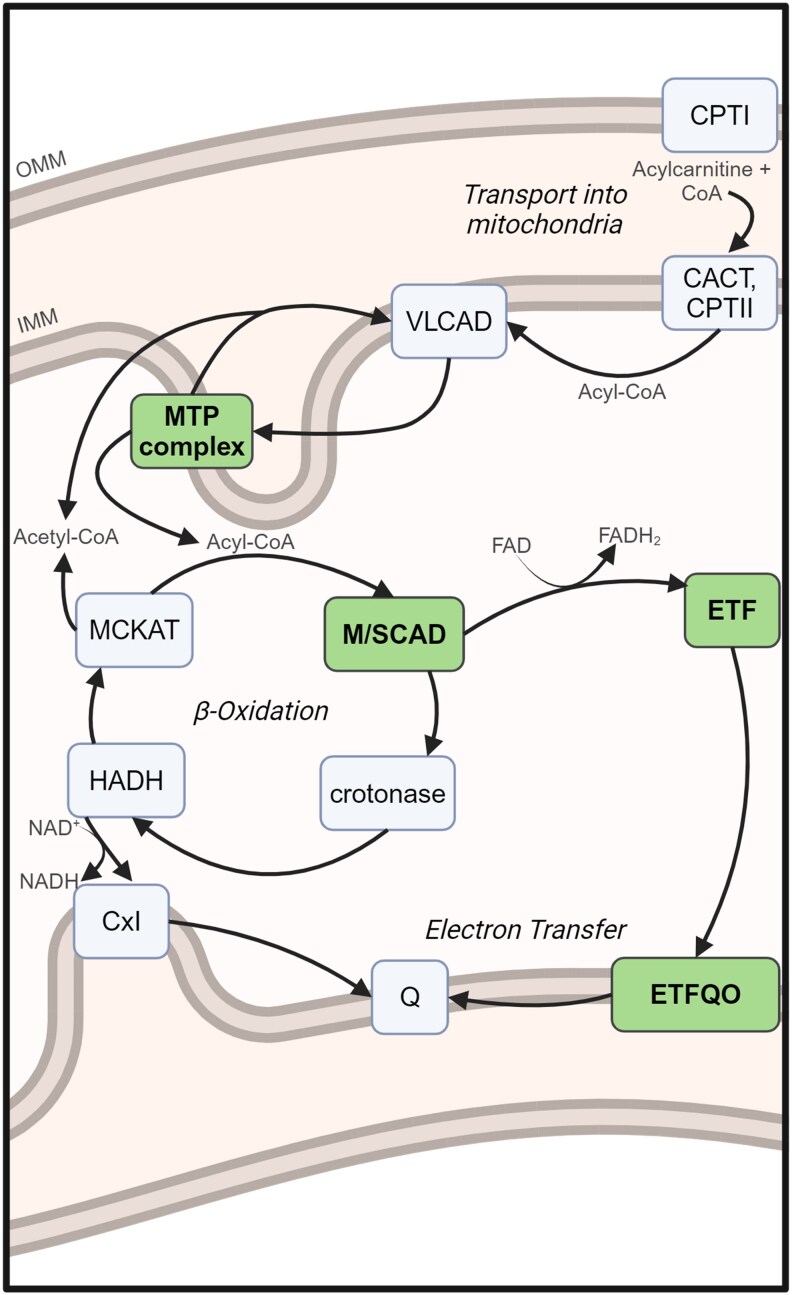 Fig. 1
Fig. 1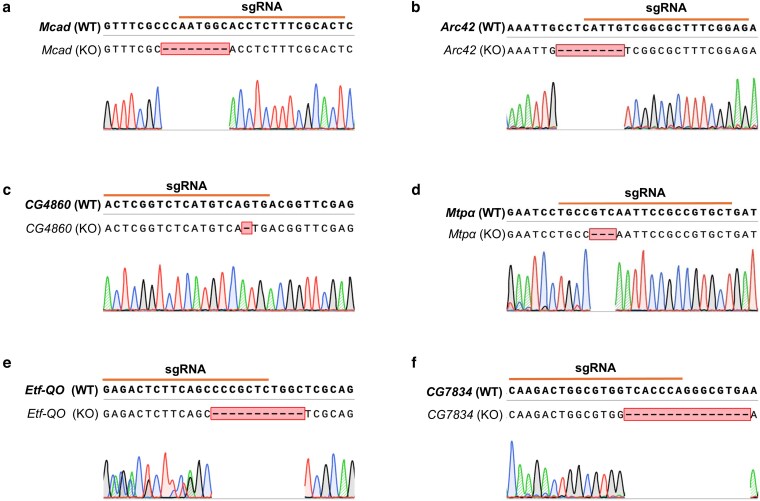 Fig. 2
Fig. 2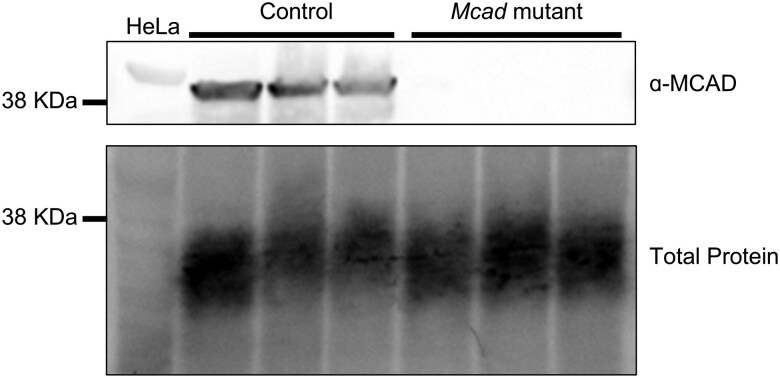 Fig. 3
Fig. 3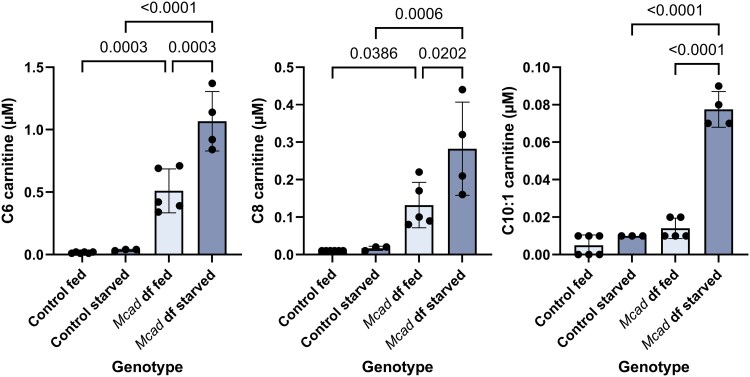 Fig. 4
Fig. 4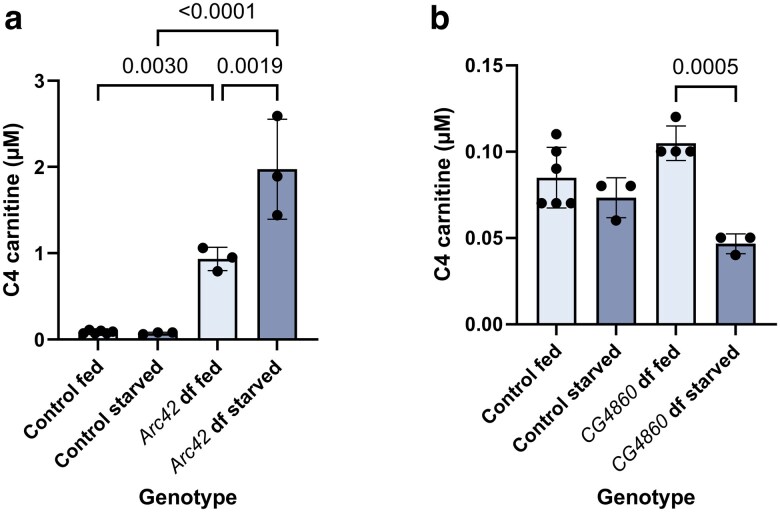 Fig. 5
Fig. 5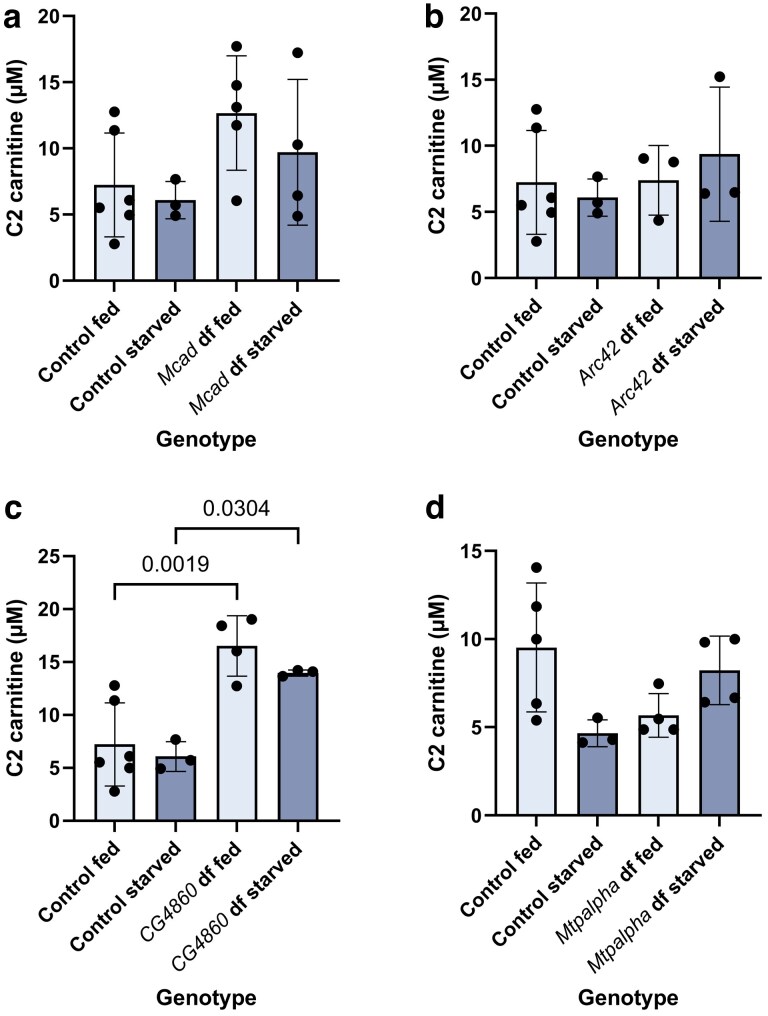 Fig. 6
Fig. 6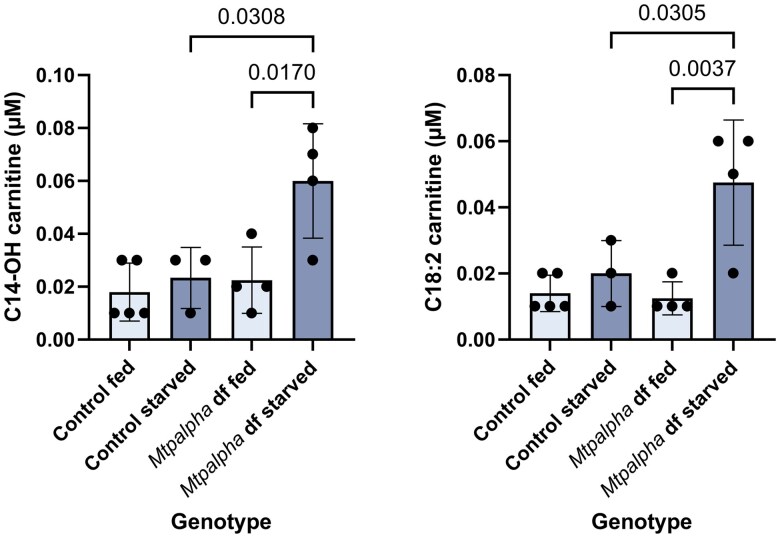 Fig. 7
Fig. 7| Human symbol | Human name | Fly symbol | Fly name |
|---|---|---|---|
| β-Oxidation | |||
|
| acyl-CoA dehydrogenase short chain |
| Activator-recruited cofactor subunit 42 |
|
| acyl-CoA dehydrogenase short chain |
| ( |
|
| acyl-CoA dehydrogenase medium-chain |
| Medium-chain acyl-CoA dehydrogenase |
|
| hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha |
| Mitochondrial trifunctional protein α subunit |
| Electron transfer | |||
|
| electron transfer flavoprotein dehydrogenase |
| Electron transfer flavoprotein-ubiquinone oxidoreductase |
|
| electron transfer flavoprotein subunit beta |
| Electron transfer flavoprotein beta subunit |
| Relevant fly target | Genotype | BDSC RRID |
|---|---|---|
|
| y[1] sc[*] v[1] sev[21]; P{y[ + t7.7] v[ + t1.8] = TKO.GS00546}attP40 | BDSC_76377 |
|
| y[1] sc[*] v[1] sev[21]; P{y[ + t7.7] v[ + t1.8] = TKO.GS03434}attP40 | BDSC_83752 |
|
| y[1] sc[*] v[1] sev[21]; P{y[ + t7.7] v[ + t1.8] = TKO.GS01855}attP40 | BDSC_79795 |
|
| y[1] sc[*] v[1] sev[21]; P{y[ + t7.7] v[ + t1.8] = TKO.GS01805}attP40 | BDSC_79768 |
|
| y[1] sc[*] v[1] sev[21]; P{y[ + t7.7] v[ + t1.8] = TKO.GS00853}attP40 | BDSC_77065 |
|
| y[1] sc[*] v[1] sev[21]; P{y[ + t7.7] v[ + t1.8] = TKO.GS00727}attP40 | BDSC_77201 |
|
| y[1] sc[*] v[1] sev[21]; P{y[ + t7.7] v[ + t1.8] = nanos-Cas9.R}attP2 | BDSC_78782 |
|
| y[1] sc[*] v[1] sev[21]; In(2LR)Gla, wg[Gla-1] PPO1[Bc]/CyO | BDSC_35781 |
|
| y[1] sc[*] v[1] sev[21]; Dr[1] e[1]/TM3, Sb[1] | BDSC_32261 |
|
| y[1] v[1]; P{y[ + t7.7] = CaryP}Msp300[attP40] | BDSC_36304 |
| Fly gene targeted | Primers (5′→3′) | |
|---|---|---|
| Forward | Reverse | |
|
| CTCTGGTCACACTGTCCATTT | CATCTGGCGGATCTGTTTCT |
|
| AGGATAATCGCTGGTGGAAAC | GCACCCAGGTACAGGTTATTC |
|
| TGGAGTTAAGCGTGTGATCG | TCCAGGCCACCATCAATTT |
|
| CGGGTCTAAAGTCCAACTTGT | CCTCAACACAACGCTCCATA |
|
| CCACATTCAAATGGCGTTCC | GAGATCCAGGCCACCAATATC |
|
| GATCAAGATCGACTCACCCAA | AGTTTGGTCTTGCTGTCCTT |
| Fly symbol | Closest human symbol | Mutation generated | Observed phenotype |
|---|---|---|---|
|
|
| 8 bp deletion, predicted frameshift | Elevated C4 acylcarnitine ( |
|
|
| 1 bp deletion, predicted frameshift | Lower C4 acylcarnitine, elevated C2 acylcarnitine |
| ( | |||
|
|
| 8 bp deletion, predicted frameshift, confirmed protein knockout | Elevated C6, C8, and C10:1 acylcarnitine ( |
|
|
| 3 bp deletion, predicted removal of one codon | Elevated C14-OH and C18:2 acylcarnitine ( |
|
|
| 11 bp deletion, predicted frameshift | Homozygous lethal |
|
|
| 15 bp deletion, predicted frameshift | Homozygous lethal |
- —CC Natural Sciences Executive Committee Research and Development
- —CC Student Collaborative Research
- —Seattle Children’s Research Institute Center for Clinical and Translational Research10.13039/100009992
- —AIS10.13039/100009064
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPeroxisome Proliferator-Activated Receptors · Lipid metabolism and biosynthesis · Mitochondrial Function and Pathology
Introduction
Inborn errors of metabolism (IEMs) are devastating genetic diseases that can manifest at any age, typically in response to physiological stress. Because IEMs account for a notable percentage of sudden infant death syndrome cases (van Rijt et al. 2016), many are included in newborn screening. Fatty acid oxidation disorders (FAODs) are a subtype of IEM caused by the inability to break down fatty acids, all of which are inherited as autosomal recessive conditions. Despite known biochemical mechanisms, there is significant variability in disease severity, and the pathophysiology is still not fully understood (Houten and Wanders 2010). Therapy is primarily dietary management and treatment of symptoms, the effectiveness of which varies by disease. Currently, FAODs are identified by characteristic acylcarnitine profiles, measured in blood by tandem mass spectrometry (Miller et al. 2021).
Fatty acids, glucose, and amino acids are the building blocks of macromolecules and major sources of energy in the body. Glucose acts as the primary source of energy through cellular respiration and glycolysis; however, when cells are depleted of glucose (e.g. stressful conditions), they rely on fatty acid metabolism. Mitochondrial fatty acid oxidation (FAO) occurs in 3 steps: (1) fatty acid transport into the mitochondria via the exchange of coenzyme A (CoA) with carnitine, (2) β-oxidation, in which acyl-CoAs are shortened by 2 carbons in a spiral pathway, and (3) electron transfer, in which the reduced electron carriers ferry their electrons to the electron transport chain (Fig. 1) (Saudubray et al. 2016).
Schematic of the mitochondrial fatty acid oxidation pathway in humans. Enzymes in this study are in bold. Some substrates and products have been omitted for simplicity. CACT, carnitine acylcarnitine translocase; CoA, coenzyme A; CPT, carnitine palmitoyltransferase; Cx I, complex I of respiratory chain; ETF, electron transfer flavoprotein; ETFQO, ETF ubiquinone oxidoreductase; FAD, flavin adenine dinucleotide; FADH2, reduced FAD; HADH, 3-hydroxyacyl-CoA dehydrogenase; IMM, inner mitochondrial membrane; MCKAT, medium-chain ketoacyl-CoA thiolase; MTP, mitochondrial trifunctional protein; M/SCAD, medium-chain/short-chain acyl-CoA dehydrogenase; NAD+, nicotinamide adenine dinucleotide; NADH, reduced NAD; OMM, outer mitochondrial membrane; Q, ubiquinone; VLCAD, very-long-chain acyl-CoA dehydrogenase. Adapted from Saudubray et al. (2016). Created in BioRender. Heimerl (2025). https://BioRender.com/o91q148.
As part of mitochondrial metabolism, FAO is relatively conserved across species, opening the possibility of studying FAODs in model organisms. Several mouse models of FAODs exist, though the mouse phenotype does not always match the human symptomology (Houten and Wanders 2010; Gaston et al. 2023). Our previous work showed that a Drosophila model of one of these deficiencies [medium-chain acyl-CoA dehydrogenase (MCAD) deficiency] recapitulated the metabolic patterns observed in human patients (Course et al. 2018). Buoyed by the similarities between human and Drosophila acylcarnitine profiles, we were interested in continuing to test putative Drosophila orthologs of human FAO genes for functional conservation.
Drosophila is one of the most genetically tractable model organisms, with high fecundity, rapid reproduction, low costs, and easily controlled environmental conditions, making them an ideal model for studying FAOD variability (Hales et al. 2015). In this study, we explore putative orthologs of human FAO genes in Drosophila to establish them as robust models of FAODs. To this end, we used CRISPR-Cas9 to mutate 6 genes in Drosophila, summarized in Table 1 and Fig. 1, then observed their resultant phenotypes. We find that while Arc42 and CG4860 are both predicted orthologs of human ACADS, the acylcarnitine profile for Arc42 loss of function mimics that of ACADS loss of function, while that of CG4860 does not. Acylcarnitine profiles also recapitulate our previous finding that Mcad is the likely ortholog of human ACADM, and show the deleterious effects of a single codon deletion in Mtpα (the predicted ortholog of human HADHA). Finally, we observe that loss of function for Etf-QO as well as for *CG7834—*predicted orthologs of human ETFDH and ETFB, respectively—is homozygous lethal in flies.
Methods and materials
Generation of Drosophila mutations
All Drosophila mutations were generated using reagents and protocols from the DRSC/Transgenic RNAi Project (TRiP) Functional Genomics Resources at Harvard Medical School (Zirin et al. 2020). Briefly, a nanos-Cas9 stock was crossed to a TRiP-KO stock containing the target sgRNA (Table 2) (Ren et al. 2013). Male F1 progeny containing both transgenes were collected and crossed to the appropriate balancer strain en masse (Table 2). The F2 progeny were collected and individually crossed to the appropriate balancer strain. Finally, the balanced F3 progeny were collected while transgenes were eliminated using a visible marker. Control lines were made in the same manner, including balancing for each target chromosome, using the fly that the sgRNAs were injected into (Table 2). All flies were obtained from the Bloomington Drosophila Stock Center (BDSC) and maintained at 25°C on Nutri-Fly Bloomington Formulation food (Flystuff) with propionic acid. FlyBase was used throughout this study (Öztürk-Çolak et al. 2024).
PCR, gel electrophoresis, and purification
To confirm mutations, PCR was performed on DNA extracted from single flies using OneTaq Quick-Load 2× Master Mix with Standard Buffer (New England Biolabs) following the manufacturer's protocol and using primers described in Table 3. To confirm successful PCR amplification, samples were run on a 1% w/v agarose gel made with 1× TAE and SYBR Safe DNA Gel Stain (Invitrogen), alongside a 1 kb Plus DNA Ladder for Safe Stains (New England Biolabs). To prepare for sequencing, PCR products were purified using a Monarch PCR and DNA Cleanup Kit (New England Biolabs), following the manufacturer's protocol.
Sanger sequencing and analysis
Purified samples were sent to Genewiz (Azenta Life Sciences) for Sanger sequencing. Sequences were assessed for quality and aligned to the wild-type Drosophila genome using Benchling (https://benchling.com). Alignment revealed deletions present in the mutant flies, all located within the sgRNA target regions. Sequencing of each sample was repeated 3 times to ensure that the same mutation was present each time. Chromatogram images used in this paper were made using SnapGene (Dotmatics).
Western blotting
To confirm protein knockout in the Mcad mutant flies, one 1–4-day old male and one 1–4-day old female fly per sample were homogenized in RIPA buffer (Thermo Scientific) at a ratio of 10 µL per fly. Samples were homogenized with pellet pestles, then centrifuged at 13,000 rpm at 4°C for 30 min to remove debris. Supernatant was run on an Any kD Mini-Protean TGX Precast Protein Gel (Bio-Rad) along with HeLa whole cell lysate (Abcam ab29545). The proteins were immunoblotted with anti-MCAD antibody at 1:500 (Abcam ab110296), followed by HRP-conjugated goat anti-mouse IgG (Jackson ImmunoResearch 115035166) at 1:5000. Total protein labeling was accomplished using the No-Stain Protein Labeling Reagent (Invitrogen), following the manufacturer's protocol. Chemiluminescent visualization was performed using SuperSignal West Pico Plus Chemiluminescent Substrate (Thermo Scientific) and imaged on an iBright CL1500 Imaging System (Invitrogen).
Acylcarnitine profiles
All flies were flipped into fresh food every 3 days and assayed at 6 days old. For the starvation cohort, flies were transferred to a vial containing only water for the final 24 h before extraction. Six flies per sample (3 female and 3 male) were homogenized in 3:1 methanol:acetonitrile (10 µL per fly). Samples were then centrifuged at 10,000 rpm for 2 min to remove debris. 45 µL of supernatant was removed and placed into a clean centrifuge tube, which was then left open in a 37°C incubator for 1 h, until all the liquid had evaporated for stability during shipping.
Sample analysis was performed similarly to previous work (Course et al. 2018). Controls (high, low, and negative) were included with each batch of acylcarnitines using 25 μL of sample. Isotope-labeled internal standards (Cambridge Isotope) in acetonitrile and 0.4% formic acid (Sigma) were added directly to each sample to a total volume of 225 μL. Samples were vortexed and centrifuged for 5 min at 13,000 rpm. Supernatant was decanted into glass reaction vials and dried for at least 45 min under nitrogen. Acylcarnitines were resuspended and derivatized in 100 μL of 3N HCl in butanol (Regis Technologies Inc.) and incubated at 50°C for 15 min. Samples were dried again under nitrogen. 200 μL of hexane was added to each vial, vortexed, then inverted to pour out hexane and remove phospholipids. Samples were dried to completion under nitrogen to ensure hexane removal. Finally, samples were resuspended in 100 μL of 80% acetonitrile and transferred to glass injection vials for analysis on a tandem mass spectrometer (Waters Xevo TQ). Injection volume was set at 10 μL. The acylcarnitine profile was collected in positive mode using precursor scanning of m/z 85. Data were acquired and quantified using MassLynx version 4.1 and NeoLynx Browser version 4.1.
Protein alignment
Protein sequences were obtained from UniProt (The UniProt Consortium 2025). Alignments were performed using Clustal Omega (Madeira et al. 2024). SUPERFAMILY protein domains were identified using InterPro (Blum et al. 2025).
Statistical analysis
Acylcarnitine profiles were statistically analyzed using One-Way ANOVA followed by Tukey's multiple comparisons. Analyses were performed using Prism version 10.4.0 for Windows (GraphPad Software, Boston, Massachusetts USA, www.graphpad.com).
Results
Identifying target genes
To determine which Drosophila genes were the predicted orthologs of 16 human FAOD genes (Saudubray et al. 2016), we used the Drosophila RNAi Screening Center (DRSC) Integrative Ortholog Prediction Tool (DIOPT; Supplementary Table 1) (Hu et al. 2011). All genes had at least 1 predicted ortholog in Drosophila, with the exception that ACADVL and ACAD9 were both predicted to be orthologs of fly CG7461; this is unsurprising, given that in humans these are considered homologous (Saudubray et al. 2016). We then cross-referenced the moderate-to-high scoring genes from this list to FlyBase (Öztürk-Çolak et al. 2024) to confirm predicted function. Finally, we cross-referenced this list to the flies carrying sgRNAs targeting these genes available through the DRSC/TRiP Functional Genomics Resources in vivo CRISPR fly stocks (Zirin et al. 2020), of which there were 13 This list included for the best predicted DIOPT score for each gene, with the exception of predicted orthologs of SLC25A20, CPTII, and NADK2; again, the predicted ortholog for ACAD9 and ACADVL was the same. We obtained 2 different lines carrying sgRNAs targeting predicted orthologs of ACADS: for CG4860 and Arc42. Ultimately, 6 lines plus 2 control lines were successfully crossed and screened for mutations. The other 7 remain untested for technical reasons. We elected to generate knockouts instead of knockdowns because many of these disorders' symptomologies are masked by residual activity of the enzyme.
Deletion mutations generated by CRISPR-Cas9
The available flies carrying sgRNAs targeting our genes of interest were crossed to a nanos-Cas9 stock, which expresses Cas9 in the germline (Ren et al. 2013). After selection and balancing, we generated 5 fly lines with early frameshift mutations, in: Mcad, Arc42, CG4860, Etf-QO, and CG7834 (Fig. 1; Table 1). Mutations were confirmed via PCR and Sanger sequencing, and all consisted of deletions ranging from 1 to 15 base pairs, all in the sgRNA target region (Fig. 2). Because the sgRNAs all targeted sequences in exon 2 and the recovered mutations were predicted to cause frameshifts, we predicted loss of function for each gene's encoded protein. Another gene, Mtpα, was also successfully mutated using this process, but the resulting 3 base pair deletion led to the removal of a single valine (Fig. 2d). Successful knockout of MCAD protein was confirmed via western blot: the MCAD protein was present in all control flies, and not in the mutant flies (Fig. 3; Supplementary Fig. 1). We performed western blots for the other mutant flies; however, commercially available antibodies were only verified to react with vertebrate proteins and did not detect the proteins in flies, thus precluding our ability to verify protein knockout.
Validation of deletions in mutated flies. Sanger sequencing chromatograms show deletion region in a) Mcad, b) Arc42, c) CG4860, d) Mtpɑ, e) Etf-QO, and f) CG7834. Barred in orange is the sgRNA target region. The first row is the wild-type sequence of the gene of interest; the second row is the aligned mutated sequence. The deletions present in the mutated flies are boxed in their respective KO sequences.
MCAD protein is absent in Mcad mutant flies. Western blotting of HeLa lysate, control fly lysate, and Mcad mutant fly lysate, probed with ɑ-MCAD antibody (top). Loading for the same blot was confirmed using a total protein label (bottom). KDa is kilodaltons. Raw images are provided in Supplementary Fig. 1.
After successfully mutating these genes in flies, we tested whether they shared functional conservation with their predicted human ortholog by acylcarnitine analysis. Acylcarnitine profiles are the primary method used to identify human FAODs. Briefly, tandem mass spectrometry quantifies a variety of acylcarnitines in a sample, and these are compared with established normal ranges for each metabolite. Each FAOD has a stereotypical acylcarnitine profile in humans (Miller et al. 2021), and we compared the Drosophila acylcarnitine profiles to those of humans with the predicted ortholog affected.
Mcad loss of function mimics that of ACADM
Mcad, previously known as CG12262, was identified in our previous work as being the likely ortholog of human ACADM (Course et al. 2018), and was re-named Mcad because of it. It was therefore expected that our new Mcad deficient flies would recapitulate the acylcarnitine profile found in human individuals with MCAD deficiency, which includes significant elevations of acylcarnitines C6, C8, and C10:1 compared with controls. This pattern was observed in our homozygous Mcad deficient flies, and, as expected, is exacerbated in starved animals vs fed (starved means water only was provided for the final 24 h before acylcarnitine extraction) (Fig. 4). The C10:1 elevation only achieved significance under the starved condition, which is consistent with the fact that it is the least abnormal metabolite in MCAD deficiency in humans. While not a novel finding, this new fly knockout data validates the previous study, and serves as a positive control for the rest of this work.
Medium-chain acylcarnitines are elevated in Mcad mutant flies. Levels of C6, C8, and C10:1 acylcarnitines in fed and starved Mcad mutant flies and controls. “Df” means “deficient.” Error bars are standard deviation. Relevant significant differences with P-values are given according to one-way ANOVA, followed by Tukey's multiple comparisons.
It is worth noting that in these and all other samples, we observed a significant elevation of C3 (propionylcarnitine) in all fed flies as compared to starved flies. This elevation is due to the propionic acid that is commonly used in fly food to prevent fungal growth (Ashburner and Roote 2007). This finding was consistent across all samples, regardless of genotype, and was not expected to affect other acylcarnitine levels (Supplementary Table 2).
Arc42 loss of function mimics that of ACADS
Two different fly genes were predicted by DIOPT to be orthologs of human ACADS: Arc42 and CG4860 (Supplementary Table 1). While CG4860 received a lower prediction score from the algorithm, they are both annotated on FlyBase as predicted orthologs of ACADS. In human individuals with SCAD (short-chain acyl-CoA dehydrogenase) deficiency, the stereotypical acylcarnitine profile is dominated by elevated C4 (butyrylcarnitine) (Miller et al. 2021). C4 was elevated in the homozygous Arc42 mutant flies, and this elevation was exacerbated in the starved condition, as expected (Fig. 5a). In contrast, we did not observe an elevation of acylcarnitine C4 in the homozygous CG4860 mutant flies as compared to controls in either condition (Fig. 5b). Instead, we observed a significantly lower concentration of C4, and a significant elevation of C2 (acteylcarnitine). This is the only genotype for which we observed an elevation in C2 (Fig. 6).
Short chain acylcarnitines are elevated in Arc42 but not CG4860 mutant flies. Levels of C4 acylcarnitines in fed and starved a) Arc42 and b) CG4860 mutant flies and controls. “Df” means “deficient.” Error bars are standard deviation. Relevant significant differences with P-values are given according to one-way ANOVA, followed by Tukey's multiple comparisons.
Acylcarnitine C2 levels across homozygous mutant flies. Shown for a) Mcad, b) Arc42, c) CG4860, and d) Mtpɑ. “Df” means “deficient.” Error bars are standard deviation. Relevant significant differences with P-values are given according to one-way ANOVA, followed by Tukey's multiple comparisons.
A single codon deletion disrupts Mtpα function
The fly gene Mtpα was predicted by DIOPT to be orthologous to human HADHA. Pathogenic variants in HADHA, as well as in HADHB, cause long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency and trifunctional protein (TFP) deficiency. In these enzyme deficiencies, acylcarnitines C16, C18, C18:2, C18:1, C14–OH, C16–OH, and C18–OH are elevated.
The deletion induced by CRISPR-Cas9 in our flies removed exactly 1 codon encoding a valine within exon 2 (Fig. 2d). Despite not being a frameshift mutation, we were still interested to see whether this change would affect protein function, especially as the amino acid at this location in humans is an isoleucine (Supplementary Figs. 2 and 3). While not identical, valine and isoleucine are both branched chain amino acids, and are structurally similar. Acylcarnitine profiles of these flies and their controls showed that C14–OH and C18:2 are significantly elevated under starvation conditions (Fig. 7).
Some long chain acylcarnitines are elevated in starved Mtpɑ mutant flies. Levels of C14-OH and C18:2 acylcarnitines in fed and starved Mtpɑ mutant flies and controls. “Df” means “deficient.” Error bars are standard deviation. Relevant significant differences with P-values are given according to one-way ANOVA, followed by Tukey's multiple comparisons.
Etf-QO and CG7834 disruptions are both homozygous lethal in Drosophila
In addition to studying 4 genes implicated in β-oxidation, we studied 2 genes involved in electron transfer: Etf-QO and CG7834. Knocking out each gene turned out to be homozygous lethal in Drosophila. We did run acylcarnitine profiles on the heterozygous Etf-QO flies, but as expected, did not observe the numerous acylcarnitine elevations that are present in humans with deficiencies in the orthologous gene (Supplementary Table 2). In humans with electron transfer deficiency, called multiple acyl-coA dehydrogenase (MAD) deficiency, we expect to see wide-ranging and severe alteration of acylcarnitines. Because our flies still have 1 functional copy of Etf-QO in this example, it is not surprising that it does not recapitulate the acylcarnitine profile of a human with an autosomal recessive and therefore homozygous disorder.
Discussion
In this study, we used CRISPR-Cas9 to generate mutations in Drosophila in 6 putative FAO genes: Mcad, Arc42, CG4860, MTPα, Etf-QO, and CG7834 (Fig. 1; Table 1). We first showed that CRISPR-Cas9 successfully removed 1–15 base pairs in each fly, leading to an early frameshift mutation and therefore a predicted knockout in 5, and the removal of 1 amino acid in 1 (Fig. 2; Table 4). In the Mcad mutant flies, we were additionally able to confirm protein knockout via western blot (Fig. 3; Supplementary Fig. 1).
We then used acylcarnitine analysis to measure the acylcarnitine levels in each of the fly mutants, as compared to controls. For the Mcad knockout flies, we observed the C6, C8, and C10:1 elevations characteristic of MCAD deficiency in humans (Fig. 4) (Course et al. 2018). For the Arc42 mutants, we observed the C4 elevation characteristic of SCAD deficiency in humans (Fig. 5a), but did not observe this pattern for CG4860 (Fig. 5b). For the MTPα mutants, we observed that just 1 amino acid deletion led to elevations in some of the acylcarnitines that are elevated in LCHAD and TFP deficiencies (Fig. 7). Finally, for the electron transfer knockouts Etf-QO, and CG7834, both strains were homozygous lethal (findings summarized in Table 4).
We will discuss each of these genes further individually. MCAD deficiency is the most common inherited FAOD (Chang et al. 2024), caused by a disruption in the gene ACADM. Individuals with MCAD deficiency experience a wide range of symptom severity, including death, usually precipitated by fasting or increased energy requirements. Acylcarnitine profiles in humans with MCAD deficiency are characterized by elevations in C6, C8, and C10:1 acylcarnitines, with the predominant peak at C8 (Miller et al. 2021). In a mouse with Acadm knocked out, these 3 acylcarnitines were also elevated, but with higher C6 and C10:1 elevations than C8 in serum (Tolwani et al. 2005). Here in Drosophila, we observe that again these 3 acylcarnitines are elevated in flies with Mcad knocked out, with the greatest elevation in C6 (Fig. 4). This observation is consistent with the acylcarnitine profiles observed for a different Mcad knockout that we had made previously (Course et al. 2018). That mutant fly was made using a different CRISPR-Cas9 technique and resulted in a different deletion, showing that the finding is reproducible. While not surprising, this result validates our finding that the previously named CG12262 is likely the ortholog of human ACADM, and acts as a kind of “positive control” for this study.
SCAD deficiency, due to its lack of severity, has been re-classified as a biochemical phenotype rather than a disease. That said, while some individuals remain asymptomatic, some infants do experience failure to thrive, hypotonia, and/or seizures. Again, there is a wide range of severity, at least partially influenced by the feeding and stress state of the individual (Wolfe et al. 2018). SCAD deficiency, caused by a disruption in the gene ACADS, is characterized biochemically in humans by an elevation in C4 acylcarnitine (Miller et al. 2021). A mouse model of SCAD deficiency arose spontaneously several decades ago, and was confirmed to have elevated C4, as well (Schiffer et al. 1989; Wood et al. 1989). More than 1 fly gene has been predicted to be the ortholog of ACADS, with the top 2 from DIOPT being Arc42 and CG4860 (Supplementary Table 1). Arc42 received a higher score, but both are annotated on FlyBase as orthologous to ACADS, so we were interested in observing both. By comparing their acylcarnitine profiles, we could see that Arc42 knockout flies exhibited the C4 elevation characteristic of humans and mice lacking functional SCAD, while CG4860 knockout flies did not (Fig. 5). Instead, we observe a significantly lower level of C4 in starved CG4860 knockout flies, which could indicate a role in SCAD synthesis. In addition, this is the only fly for which we observe a modest but significant elevation of C2, which could be related to a role in ketone utilization (Fig. 6).
HADHA, along with HADHB, encodes a protein involved in the enzymatic complex mitochondrial trifunctional protein (MTP, Fig. 1). Biallelic pathogenic variants in this gene lead to LCHAD deficiency and TFP deficiency in humans. Both of these deficiencies can result in a variety of symptomologies and severities (Prasun et al. 2022). In addition, carriers of pathogenic variants in either gene are at a high risk of HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome during pregnancy when carrying an affected fetus (Saudubray et al. 2016). The characteristic acylcarnitine profile in affected humans shows elevations in long-chain acylcarnitines C16, C18:2, C18:1, C18, C14–OH, C16–OH, and C18–OH. In mice, knockout of the orthologous gene Hadha is neonatal lethal and significantly elevated serum acylcarnitines were C14, C16:1, C16, C18:2, and C18:1 (Ibdah et al. 2001). Another mouse model in which a common pathogenic variant, G1528C, was knocked into the mice using CRISPR-Cas9 resulted in phenotypes relevant to LCHAD deficiency, including blood plasma elevations in C16:1, C16, C18:2, C18, C16–OH, C18:1–OH, and C18–OH.
In flies, when the putative orthologous gene, Mtpα, was knocked out using the ends-out gene targeting technique, flies were not early-stage lethal as the mouse was (Kishita, Tsuda, and Aigaki 2012). This study found significant increases in the relative amounts of virtually all saturated (C18–C10) and unsaturated (C18–C12) fatty acid acylcarnitines and hydroxyacylcarnitines (C18–C12) in these flies’ vs controls. We sought to recapitulate this data here with our method of CRISPR-Cas9 knockout, but our CRISPR-Cas9 led to the exact removal of 1 amino acid—a valine—encoded in exon 2 (Fig. 2d). This valine aligns with an isoleucine in the human protein (Supplementary Figs. 2 and 3); because they are both branched chain amino acids, they are considered to affect the protein structure similarly. Intriguingly, this missense mutation still affected the acylcarnitine profile: C14–OH and 18:2 were significantly elevated in the starvation condition (Fig. 7). This finding suggests that the single valine deletion had a small but significant effect on overall enzyme function.
In addition to the above genes involved in β-oxidation, we also knocked out 2 genes predicted to be involved in electron transfer: Etf-QO and CG7834. Intriguingly, both mutant strains were homozygous lethal. We did perform acylcarnitine analysis on the Etf-QO heterozygotes, and unsurprisingly found no significant results (Supplementary Table 2). Etf-QO is predicted to be the fly ortholog of human ETFDH, while CG7834 is predicted to be the fly ortholog of human ETFB. Biallelic pathogenic variants in either cause MAD deficiency, which can exhibit a wide range of severity as in the β-oxidation disorders, but often more severe and including death in early infancy. The fact that knockout of either electron transfer gene caused homozygous lethality in our flies is therefore not inconsistent with the human deficiencies, in the sense that humans show severe symptomology. On the other hand, it is not typically lethal in humans until a stress event, which suggests that this loss of function is more deleterious in flies.
In Etf-QO mutants made previously using ethyl methanesulfonate, the flies were homozygous lethal, which is consistent with what we observed here (Alves et al. 2012). Similarly, in C. elegans, loss of the ETFDH ortholog, let-721, is maternal effect lethal (Chew et al. 2009). In zebrafish, loss of the ortholog etfdh leads to acylcarnitine elevations in C4, C5, C6, C8, C14, C16, and C18 (Song et al. 2009). This overlaps with characteristic profile of MAD deficiency in humans: unsaturated acylcarnitines C4–18, saturated C14–18, and C3-DC are all expected to be elevated (Miller et al. 2021). Less work has been done on models of ETFB. The fact that loss of the putative fly ortholog, CG7834, exhibits the same homozygous lethal phenotype as that of loss of Etf-QO is consistent, and suggests that this gene is involved in the same pathway. Conditional knockout fly models would need to be made to study the effects of knocking out these genes in adult flies.
Overall, this work summarizes the creation and characterization of several fly lines in which putative orthologs of human FAO genes have been mutated. We show that acylcarnitine analysis is an effective way to observe FAO changes in flies. With proper validation, flies are a good model for studying mitochondrial FAO and its related disorders. Robust animal models of these disorders, in turn, will enable the study of their progression, intervention, and perplexing variability.
Supplementary Material
jkaf139_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alves E, Henriques BJ, Rodrigues JV, Prudêncio P, Rocha H, Vilarinho L, Martinho RG, Gomes CM. 2012. Mutations at the flavin binding site of ETF: QO yield a MADD-like severe phenotype in Drosophila. Biochim Biophys Acta. 1822(8):1284–1292. doi:10.1016/j.bbadis.2012.05.003.22580358 · doi ↗ · pubmed ↗
- 2Ashburner M, Roote J. 2007. Maintenance of a Drosophila laboratory: general procedures. Cold Spring Harb Protoc. 2007(3):pdb.ip 35. doi:10.1101/pdb.ip 35.21357030 · doi ↗ · pubmed ↗
- 3Blum M, Andreeva A, Florentino LC, Chuguransky SR, Grego T, Hobbs E, Pinto BL, Orr A, Paysan-Lafosse T, Ponamareva I, et al 2025. Inter Pro: the protein sequence classification resource in 2025. Nucleic Acids Res. 53(D 1):D 444–D 456. doi:10.1093/nar/gkae 1082.39565202 PMC 11701551 · doi ↗ · pubmed ↗
- 4Chang IJ, Lam C, Vockley J. 2024. Medium-chain acyl-coenzyme A dehydrogenase deficiency. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. Gene Reviews®. Seattle (WA): University of Washington, Seattle. http://www.ncbi.nlm.nih.gov/books/NBK 1424/.20301597 · pubmed ↗
- 5Chew DS, Mah AK, Baillie DL. 2009. Characterizing the transcriptional regulation of Let-721, a Caenorhabditis elegans homolog of human electron flavoprotein dehydrogenase. Mol Genet Genomics. 282(6):555–570. doi:10.1007/s 00438-009-0485-z.19774399 · doi ↗ · pubmed ↗
- 6Course MM, Scott AI, Schoor C, Hsieh C-h, Papakyrikos AM, Winter D, Cowan TM, Wang X. 2018. Phosphorylation of MCAD selectively rescues PINK 1 deficiencies in behavior and metabolism. Mol Biol Cell. 29(10):1219–1227. doi:10.1091/mbc.E 18-03-0155.29563254 PMC 5935071 · doi ↗ · pubmed ↗
- 7Gaston G, Babcock S, Ryals R, Elizondo G, De Vine T, Wafai D, Packwood W, Holden S, Raber J, Lindner JR, et al 2023. A G 1528 C hadha knock-in mouse model recapitulates aspects of human clinical phenotypes for long-chain 3-hydroxyacyl-Co A dehydrogenase deficiency. Commun Biol. 6(1):1–12. doi:10.1038/s 42003-023-05268-1.37644104 PMC 10465608 · doi ↗ · pubmed ↗
- 8Hales KG, Korey CA, Larracuente AM, Roberts DM. 2015. Genetics on the fly: a primer on the Drosophila model system. Genetics. 201(3):815–842. doi:10.1534/genetics.115.183392.26564900 PMC 4649653 · doi ↗ · pubmed ↗