Impact of Bulky Substituents on the Singlet Arylnitrene Ring Enlargement
Holger F. Bettinger

TL;DR
This paper explores how bulky chemical groups affect the formation of certain reactive molecules called arylnitrenes and their transformation into didehydroazepines.
Contribution
The study compares computed energy surfaces to show that bulky substituents do not significantly alter the formation rates of didehydroazepines.
Findings
Bulky substituents like hydrindacene do not significantly change didehydroazepine formation rates.
Didehydroazepines may act as intermediates in forming stable triplet arylnitrenes.
Triplet arylnitrenes can be isolated from aryl azide photodecomposition.
Abstract
Substituents of the hydrindacene type allow the isolation of crystalline stable triplet arylnitrenes from photodecomposition of the corresponding aryl azides, while phenyl azide is known to produce singlet phenylnitrene that reacts to didehydroazepine via benzazirine. Comparison of computed potential energy surfaces for ring enlargement of singlet arylnitrenes suggests that bulky groups of the hydrindacene type should not significantly change the rates of formation of didehydroazepines compared to phenylnitrene. Thus, didehydroazepines could be trappable intermediates en route to stable triplet arylnitrenes.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
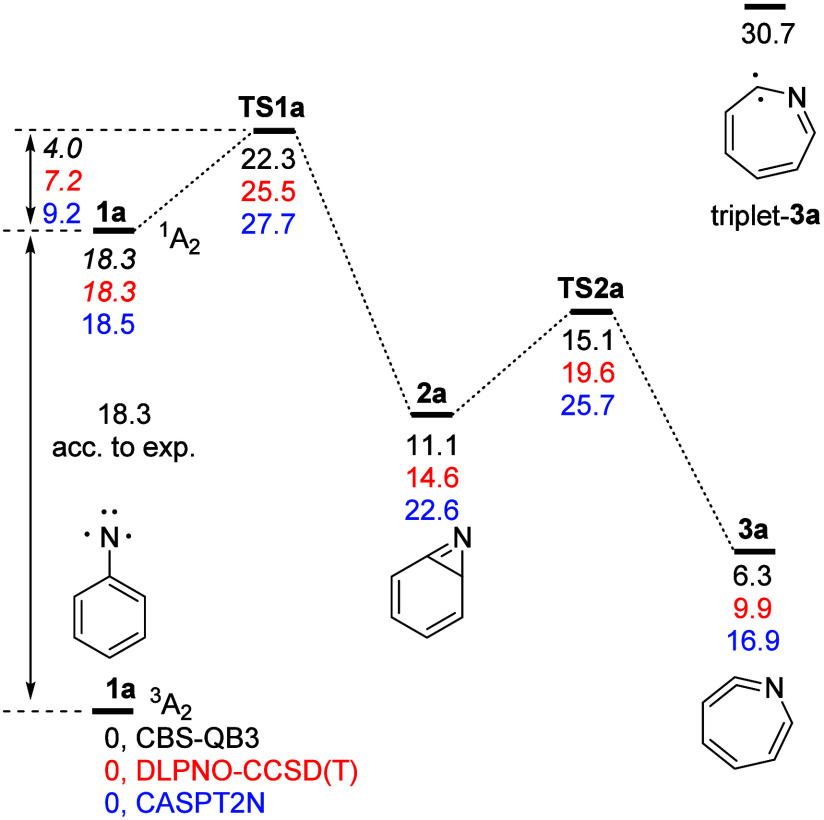 Figure 1
Figure 1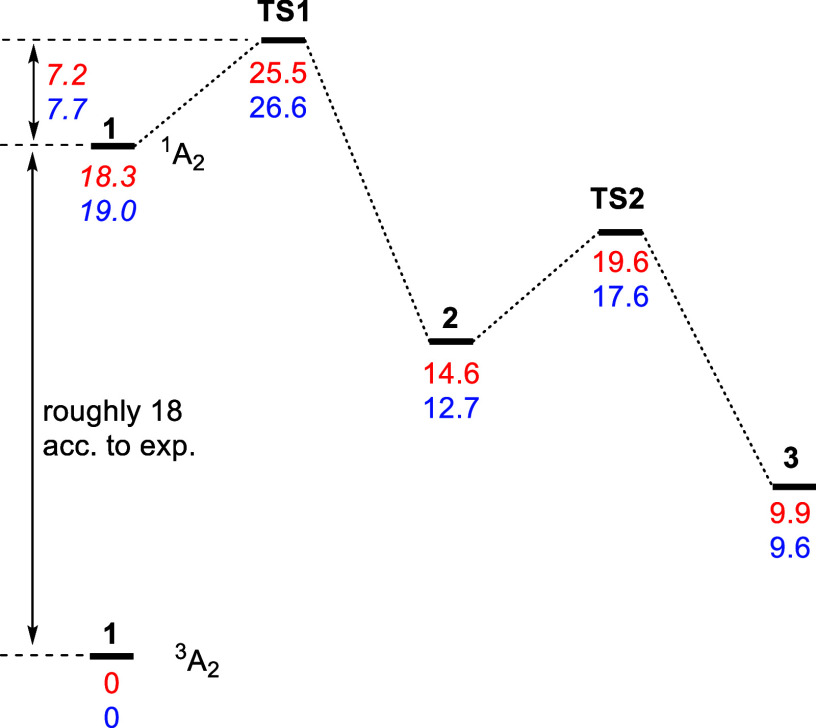 Figure 2
Figure 2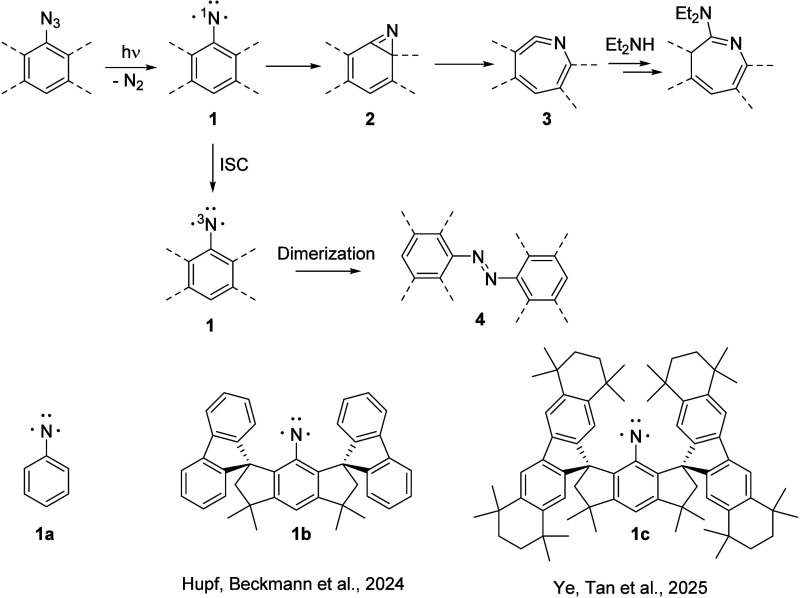 Figure 3
Figure 3 Figure 4
Figure 4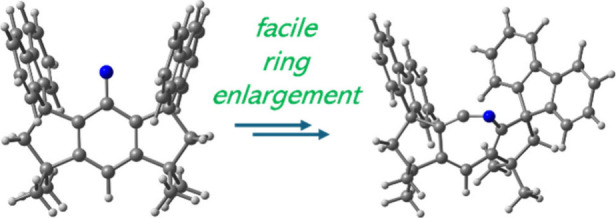 Figure 5
Figure 5- —Deutsche Forschungsgemeinschaft10.13039/501100001659
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsChemical Reactions and Mechanisms · Synthesis and Catalytic Reactions · Molecular Junctions and Nanostructures
Phenylnitrene (1a), generated photochemically from phenyl azide, is known to quickly isomerize via a benzazirine intermediate (2a) to didehydroazepine (also called ketenimine, 3a) that typically undergoes follow-up reactions (Scheme).? The reaction of 2 to 3 can proceed by heavy-atom tunneling at 10 K, but due to the small barrier, it is fast at room temperature. ?−? ? The recent reports ?,? of kinetically stabilized crystalline triplet arylnitrenes (1b, 1c; Scheme) pose the question as to why the photolysis of the corresponding azide precursor in solution at room temperature does not result in singlet arylnitrene rearrangement but rather in formation of the stable triplet in medium to high yields (52–80%). The bulky groups of 1b and 1c preclude the typically observed dimerization of triplet arylnitrenes to azobenzenes 4.
The barrier for formation of benzazirine 2a from phenylnitrene is E a = 5.6 ± 0.3 kcal/mol (A = 10^13.1 ± 0.3^ s^–1^).? Consistent with theory,? 2,6-dimethyl substitution increases the barrier for benzazirine formation (2,6-dimethyl phenylnitrene: E a = 7.0 ± 0.3 kcal/mol, A = 10^13.0 ± 0.3^ s^–1^) according to laser flash photolysis (LFP).? In contrast, 2,4,6-tri-tert-butylphenylnitrene could not be observed by LFP due to its very short lifetime, and ring opening of the benzazirine to the didehydroazepine becomes the rate-determining step (E a = 7.4 ± 0.2 kcal/mol, A = 10^12.6 ± 0.2^ s^–1^) of the rearrangement.? As the barrier for benzazirine formation is obviously quite low for 2,4,6-tri-tert-butylphenylnitrene, it may be expected that for the structurally related photogenerated singlet arylnitrenes 1b and 1c the ring enlargement should be competitive with intersystem crossing (ISC) to the triplet ground state. In this scenario, the formation of the didehydroazepine isomers 3b and 3c that are stable toward bimolecular degradation reactions due to the bulky substituents is anticipated. But no evidence for their formation was observed. ?,?
Here we compare the potential energy surface (PES) for the isomerization of the arylnitrene 1b investigated by Hupf, Beckmann, and co-workers? to that of parent phenylnitrene 1a using density functional theory (M06–2X? for geometry optimization) and DLPNO–CCSD(T) ?−? ? as implemented in Orca? for single point energy evaluations (see Supporting Information for computational details). As singlet arylnitrenes are open-shell systems (1a: ^1^A_2_ electronic state), we give the energy of the transition states and the singlet products relative to triplet arylnitrene (1a: ^3^A_2_ electronic state), and employ the experimental ?,? triplet-singlet energy gap of 1a (18.33 ± 0.69 kcal/mol) to fix the energy of the singlet state on the PES. In order to judge the performance of DLPNO–CCSD(T)//M06–2X, we employed high-level computations of the complete basis set extrapolation scheme type (CBS-QB3) ?,? for parent phenylnitrene 1a and its rearrangement to didehydroazepine 3a, and also compare to previous CASPT2N//CASSCF? computations.
First, we note that the CBS-QB3 and DLPNO–CCSD(T) methods give barriers of 4–7 kcal/mol for benzazirine 2a formation from singlet phenylnitrene 1a via TS1a that are in quite good agreement with the experimental value (E a = 5.6 ± 0.3 kcal/mol, Figure).? Previous (symmetry-broken) DFT? as well as CASPT2N? approaches arrived at a too high barrier, the latter due to an overstabilization of the phenylnitrene ^1^A_2_ state.? The impact of 2,6-dimethyl and 2,4,6-tri-tert-butyl substitution at the DLPNO–CCSD(T) level on the barrier of benzazirine formation qualitatively follows the experimental observations: compared to parent phenylnitrene 1a, methyl substituents increase the barrier for benzazirine formation by 1.2 kcal/mol (exp.? by 1.8 kcal/mol) taking into account the decrease of the triplet-singlet energy gap by 1.7 kcal/mol computed earlier at the CASPT2 level.? On the other hand, tert-butyl substituents decrease the barrier by 7.3 kcal/mol assuming that the singlet–triplet energy gap decreases by 2.4 kcal/mol due to 2,4,6-tri-tert-butyl substitution as computed earlier at the CASPT2 level.? Most importantly, the nitrene could not be observed by LFP experiments, indicating a very low barrier for benzazirine formation.? These data show that the DLPNO–CCSD(T) method can qualitatively describe the impact of the substituents correctly.
Second, we find that didehydroazepine 3a is higher in energy than ^3^A_2_ phenylnitrene 1a by around 6–10 kcal/mol. This is as much as 10 kcal/mol less than the 16.9 kcal/mol obtained at the CASPT2N level.? The employed extrapolation method relies on CCSD(T) energies, and T_1_ diagnostics do not give any indication for problems in the CCSD treatment of triplet phenylnitrene 1a (T_1_ = 0.02) or didehydroazepine 3a (T_1_ = 0.01). We thus tend to assign a lower energy to 3a. A consequence of this relatively lower energy of didehydroazepine 3a is that the rearrangement of singlet phenylnitrene 1a to didehydroazepine 3a is more exothermic (−8 to −12 kcal/mol) than concluded previously? (−1.6 kcal/mol).
The PES for parent and bulky phenylnitrene (Figure) was computed using the DLPNO–CCSD(T)/cc-pVTZ//M06–2X/6–311+G(d,p)
- ZPVE method. The triplet-singlet energy gap of bulky system 1b is expected to not differ much from that of 1a. This seems reasonable as quite similar D values were observed by EPR spectroscopy for 1a and 1c. ?,? Indeed, symmetry-broken UM06–2X computations arrive at triplet-singlet energy gaps of 18.4 kcal/mol (exp. ?,? 18.3 kcal/mol) and 19.0 kcal/mol for 1a and 1b, respectively, further supporting the assumption of similar ST gaps. Using the UM06–2X ST gap, the barrier for the formation of the benzazirine isomer from 1b is larger by 0.4 kcal/mol than that for parent 1a. Although we cannot give a better estimate of the singlet–triplet energy gap, we conclude that the barrier for benzazirine formation is likely similar to that of the parent system. Both benzazirine 2b and didehydroazepine 3b are slightly more stable relative to the singlet nitrene 1b than in the case of the parent, slightly increasing the thermodynamic driving force for rearrangement.
Most importantly, there is no indication that the barriers for didehydroazepine formation are significantly higher than those for parent phenylnitrene. Hence, it is expected that formation of singlet nitrene 1b upon photolysis should similarly result in rapid isomerization to the didehydroazepine 3b isomer. We suggest that the photolysis be performed in the presence of small nucleophiles such as diethylamine, as this is known to be able to trap didehydroazepines readily. This would allow the identification of a rearrangement mechanism also in the case of the bulky substituted arylnitrenes 1b and 1c.
Should the bulky systems undergo rearrangement as predicted, then high-yield formation of the triplet nitrenes 1b (or 1c) needs to be accounted for. An equilibration of 3 and 1 has been invoked before to explain azobenzene formation (by ISC and triplet arylnitrene dimerization). ?,? Such an equilibrium lies far on the side of 3, but with barriers of 16.8 kcal/mol (3b) and 15.4 kcal/mol (3a) formation of singlet 1 and its relaxation to the triplet state are conceivable at room temperature. It would be very interesting to run the photolysis at lower temperatures. The proposal by Schuster and co-workers? of an ISC from the singlet didehydroazepine to its triplet state (an iminocarbene) must be ruled out as this is 24.4 kcal/mol (CBS-QB3) above the singlet state (triplet-3a in Figure). An alternative explanation for the experimental observation of dominant triplet arylnitrene formation could be a photophysical mechanism that involves photosensitization by the fluorene-type large π systems of the azide and direct formation of the triplet nitrene. The details pertaining to such a process were investigated computationally for phenyl azide by Soto and Otero.?
In summary, the computations show that the rearrangements of the singlet arylnitrenes 1a and 1b share similar PES. This suggests that the photolysis of the corresponding aryl azide could result in fast formation of the didehydroazepine isomer after N_2_ extrusion, like the case of phenyl azide, provided that singlet phenylnitrene is involved as a reactive intermediate. We suggest that trapping experiments, e.g. with diethylamine, should be done to investigate the possible formation of the didehydroazepine. The computed PESs indicate that the equilibration of didehydroazepine and singlet arylnitrene is in principle feasible at room temperature. An alternative explanation for the formation of triplet nitrene could be an intramolecular photosensitization involving the organic π system of the fluorene units that ultimately avoids population of the singlet nitrene state.
Computational Methods
All structures were fully optimized using the M06–2X functional in conjunction with the 6–311+G(d,p) basis set.? Computation of harmonic vibrational frequencies confirmed the nature of stationary points as minima or transition states. For singlet nitrenes and TS1 the spin-symmetry-broken approach was used. In addition, the CBS-QB3 ?,? method was employed to compute the energy of stationary points for the rearrangement of parent phenylnitrene. The CBS-QB3 geometry and vibrational frequencies of TS1a were computed using the spin-unrestricted UB3LYP method, while the higher-level computations were done using the spin-restricted approach. The M06–2X and CBS-QB3 computations were performed using Gaussian 16.? The M06–2X/6–311+G(d,p) structures were employed for single point energy evaluation using the domain based local pair-natural orbital (DLPNO) approximation (with TightPNO setting along with the “usefullLMP2guess false” option) to coupled-cluster with singles, doubles, and a perturbative estimate of triples excitations, DLPNO–CCSD(T), ?−? ? as implemented in Orca 6? using the spin-restricted approach for all species except for triplet nitrenes that were treated with spin-unrestricted approximation. The cc-pVTZ? basis set along with the recommended RI and RIJK fitting bases was used. ?,? The DLPNO–CCSD(T)/cc-pVTZ energies were corrected for zero-point vibrational energies computed at M06–2X/6–311+G(d,p).
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Gritsan N. P.Platz M. S.Kinetics, Spectroscopy, and Computational Chemistry of Arylnitrenes Chem. Rev.20061063844386710.1021/cr 040055+16967923 · doi ↗ · pubmed ↗
- 2Nunes C. M.Eckhardt A. K.Reva I.Fausto R.Schreiner P. R.Competitive Nitrogen versus Carbon Tunneling J. Am. Chem. Soc.201914136143401434810.1021/jacs.9b 0686931423776 · doi ↗ · pubmed ↗
- 3Nunes C. M.Reva I.Kozuch S.Mc Mahon R. J.Fausto R.Photochemistry of 2-Formylphenylnitrene: A Doorway to Heavy-Atom Tunneling of a Benzazirine to a Cyclic Ketenimine J. Am. Chem. Soc.201713948176491765910.1021/jacs.7b 1049529112415 · doi ↗ · pubmed ↗
- 4Inui H.Sawada K.Oishi S.Ushida K.Mc Mahon R. J.Aryl Nitrene Rearrangements: Spectroscopic Observation of a Benzazirine and Its Ring Expansion to a Ketenimine by Heavy-Atom Tunneling J. Am. Chem. Soc.201313528102461024910.1021/ja 404172 s 23795602 · doi ↗ · pubmed ↗
- 5Janssen M.Frederichs T.Olaru M.Lork E.Hupf E.Beckmann J.Synthesis of a stable crystalline nitrene Science 2024385670631832110.1126/science.adp 496338870274 · doi ↗ · pubmed ↗
- 6Wang D.Chen W.Chen H.Chen Y.Ye S.Tan G.Isolation and characterization of a triplet nitrene Nat. Chem.2025171384310.1038/s 41557-024-01669-939562811 · doi ↗ · pubmed ↗
- 7Gritsan N. P.Zhu Z.Hadad C. M.Platz M. S.Laser Flash Photolysis and Computational Study of Singlet Phenylnitrene J. Am. Chem. Soc.199912161202120710.1021/ja 982661 q 11456816 · doi ↗ · pubmed ↗
- 8Karney W. L.Borden W. T.Why Does o-Fluorine Substitution Raise the Barrier to Ring Expansion of Phenylnitrene?J. Am. Chem. Soc.1997119143347335010.1021/ja 9644440 · doi ↗