A systems biology framework integrating cross-species transcriptomics and PPI networks for Xylella fastidiosa resistance gene identification
Aparna S. Balan, Giorgia Tranchina, Floriana Bonanno, Tiziano Caruso, Francesco Paolo Marra, Claudio Di Vaio, Annalisa Marchese

TL;DR
This study identifies 18 conserved genes involved in plant resistance to Xylella fastidiosa, a harmful bacterium, by analyzing gene activity across four plant species.
Contribution
A novel core resistance network of 18 conserved genes against Xylella fastidiosa was identified through cross-species transcriptomics and PPI networks.
Findings
18 conserved genes and 1852 divergent expression patterns were identified across four plant species.
The genes are involved in structural reinforcement, stress signaling, antimicrobial compound production, and resource optimization.
Protein-protein interaction networks revealed immune hubs like BAK1, WRKY33, and WRKY40 with new connections to proteases and ubiquitin components.
Abstract
Xylella fastidiosa, a highly pathogenic, xylem-limited, gram-negative bacterial species, represents a significant threat to many plant species, including olive, almond, grapevine, and alfalfa. Through cross-species transcriptomic analysis of Olea europaea, Prunus dulcis, Vitis vinifera, and Medicago sativa, we identified a novel core resistance network consisting of 18 conserved genes against Xylella fastidiosa, alongside 1852 divergent expression patterns. These common genes may play a crucial role in orchestrating a multi-layered plant defense response, enabling (1) structural reinforcement as well as facilitating cuticular wax biosynthesis (KCS11 and KAS1); (2) stress signaling mediated by hormonal crosstalk involving jasmonic acid (JA), salicylic acid (SA), and abscisic acid (ABA) mediated by the genes AOS and CYP707A4, alongside calcium signaling through ACA12 gene; (3)…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
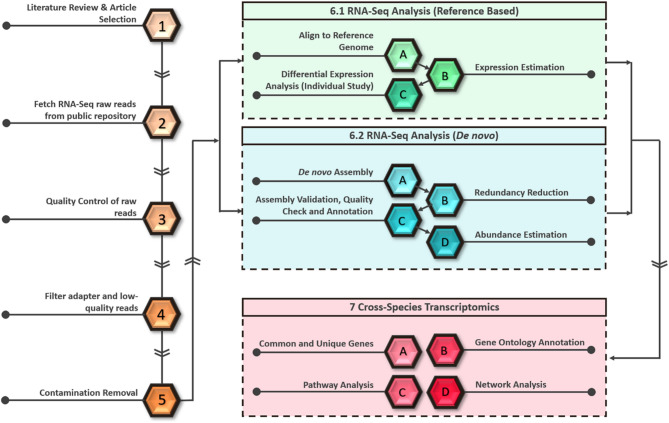 Figure 1
Figure 1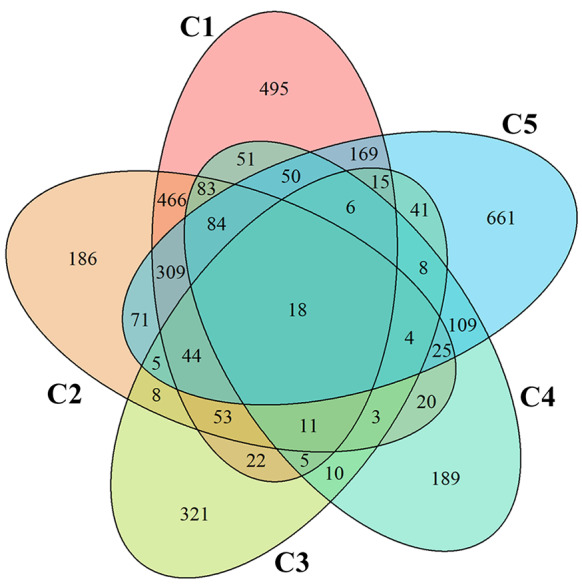 Figure 2
Figure 2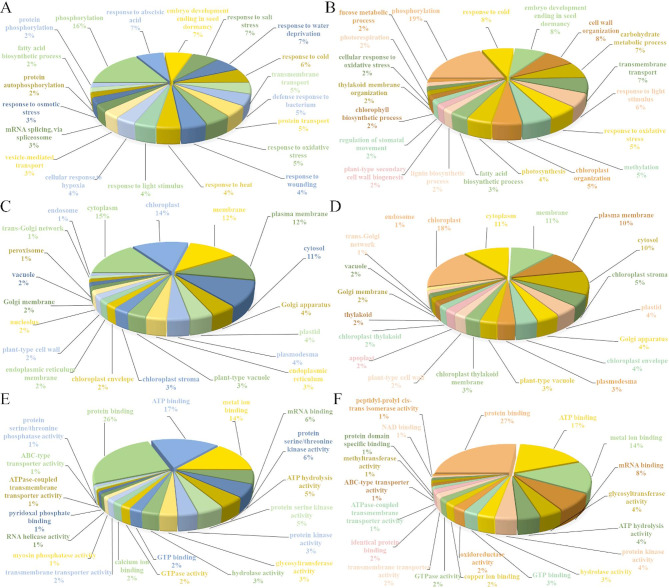 Figure 3
Figure 3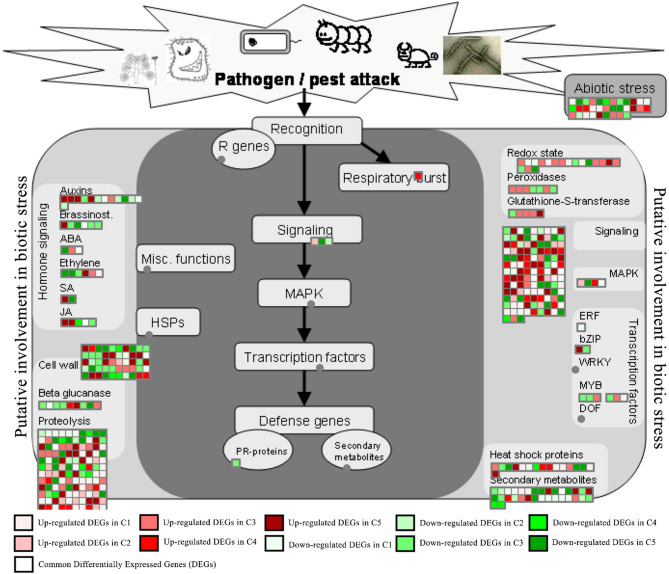 Figure 4
Figure 4 Figure 5
Figure 5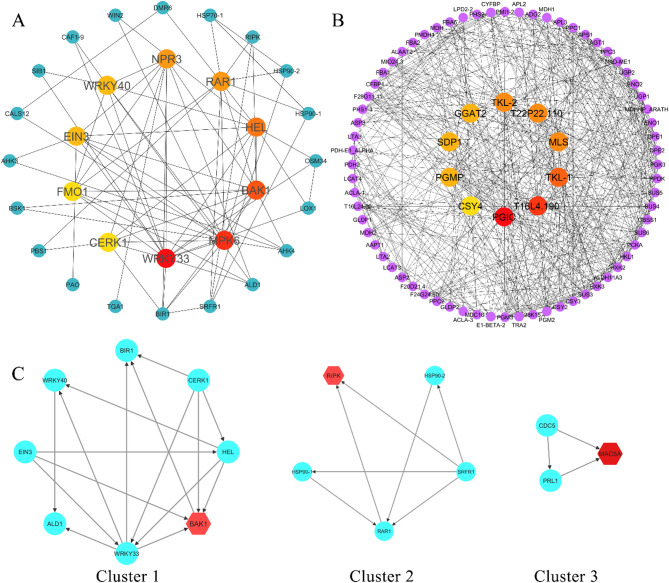 Figure 6
Figure 6- —National Grant MIPAAF Decreto n. 419161 - 13/09/2022
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhytoplasmas and Hemiptera pathogens · Cocoa and Sweet Potato Agronomy · Insect-Plant Interactions and Control
Introduction
Xylella fastidiosa (Xf) a prominent plant pathogen transmitted by xylem-sap-feeding insects, is widely known for causing significant damage to various crops on a global scale. Xf is a genetically diverse species that comprises three primary subspecies: fastidiosa, multiplex, and pauca [24]. Other subspecies such as sandyi, morus, and tashke have been suggested [19, 87, 89]. The ability of this pathogen to colonize different plant species and adapt to diverse environmental conditions contributes to its resilience and persistence within agricultural landscapes. Once a host gets infected by this bacterium it successfully colonizes inside the xylem vessels with its ability to alternate between motile cells and attachment-driven biofilm formation facilitates. The pathogen forms a significant bacterial load in the xylem vessels by moving efficiently within the vessels thereby increasing the likelihood of being acquired by insect vectors. For growth and survival, the bacterium acquires xylem sap, which contains water, minerals, organic acids, amino acids, alcohols, sugars, osmolytes, and vitamins, to satisfy all its nutritional requirements [52]. The blocked xylem vessels disrupt the flow of water along with the dissolved minerals from roots to leaves resulting in dehydration stress and nutrient shortage, adversely affecting the plant growth and development and leading to the death of the host plant.
The Xf bacterium has been reported in 40 countries across five continents, with the largest number of affected nations being in Europe, accounting for 19 of these countries. The bacterium threatens 570 host plants, seven of which are classified as major hosts, including olive, almond, grape, sweet orange, and two varieties of coffee. A total of 50 known vectors can transmit the bacterium of which 26 vectors are marked as potential (https://gd.eppo.int/taxon/XYLEFA).
The introduction of this bacterium into Europe occurred through global trade networks, resulting in the manifestation of Olive Quick Decline Syndrome (OQDS) in olive trees, first detected in 2013 in Apulia, Italy. The spread of the strain Xf subsp. pauca strain CoDiRO, ST53, in olive can occur through propagation material, but in nature, it is mainly transmitted by xylem-sap-feeding insects, such as Philaenus spumarius L. (spittlebug) [78, 84, 85]. The typical and most frequent symptoms are leaf scorching, with drying at the apical and/or marginal parts of the blade, drying of the canopy initially affecting isolated branches and then entire branches and/or the entire plant, and internal browning of the wood at various levels of the younger branches, branches, and trunk [38]. These symptoms are very similar to those caused by water stress. The bacterium Xylella fastidiosa has been included in the list of quarantine organisms of the European Union (Annex I AI of Council Directive 2000/29/EC). The infection of olive trees resulted in the death of millions of trees. The bacterium has since spread to approximately 54,000 hectares of olive trees in Apulia, raising considerable concern across the Mediterranean basin and advancing towards Central Italy [4]. The economic implications of this infection are substantial, with potential costs to Italy estimated at €5.2 billion over the next 50 years [11].
To date, no effective treatment has been identified for the control of Xf. A decade of research started from European Food Safety Authority and conducted by many researchers demonstrated that only the cvs. Leccino and Favolosa^®^ (FS17) exhibit a degree of tolerance to ST53 infections, characterized by milder symptoms and reduced bacterial populations in comparison to more susceptible cultivars such as ‘Cellina di Nardò’ and ‘Ogliarola salentina’ [51, 76, 90].Two subspecies of Xf - namely subsp. multiplex sequence type (ST) 6 and subsp. pauca - adversely affect almond production, causing almond Leaf Scorch Syndrome (ALS), leading to reduced yields and economic losses [24, 49]. Xf also attacks grapes, causing Pierce’s Disease with symptoms of leaf chlorosis, wilting, and diebacks in infected plants, which poses a significant threat to European viticulture especially the Mediterranean region [116]. The estimated product loss for grape production in European Mediterranean countries is approximately 0.71 billion USD [9].
The development of Next Generation Sequencing (NGS) technology has transformed genome research, allowing the sequencing of large numbers of genomes and transcripts in a short time, addressing the growing demand for deeper understanding of genetic mutations and their links to diseases [33, 37]. Nowadays, a huge amount of RNA-Seq data generated by the high throughput sequencing technique is deposited in the public repository, which can be reused to generate new knowledge and scientific findings through cross-species transcriptomics analysis approach by integrating and jointly analyzing these datasets [37, 43, 104]. The capability of cross-species transcriptomics analysis approach to combine data from multiple independent studies increases statistical power, enhances generalizability, and uncovers broader patterns or trends, which in turn pinpoints similarities, validates findings, and uncovers gene expression signatures associated with specific biological conditions, diseases, or treatments. Appropriate bioinformatic tools, techniques, and algorithms significantly enhance the cross-species transcriptomics analysis by providing ample support during the data integration, normalization, interpretation, and visualization processes. By taking advantage of modern early detection techniques and planning selection and molecular-assisted breeding programs, bioinformatics plays a crucial role in safeguarding plants [66, 86]. By sequencing the genomes of different plant varieties, researchers can identify and develop disease-resistant cultivars, with a particular focus on local genetic resources that harbour resilience genes [47]. Transcriptomics, which involves studying gene expression patterns, further enhances the understanding of plant defense mechanisms against biotic stress conditions. It also aids in the discovery of target genes that can be utilized in novel breeding techniques like genome editing [6].
Even though individual RNA-Seq studies have uncovered key findings into species-specific responses, integrating data from multiple plant species is essential for identifying both conserved and unique gene expression patterns across various host plants. Cross-species analysis also improves the robustness of findings, minimizing biases from individual studies and increasing the applicability of results. Thus, in this study, we examined RNA-Seq data from four species - Olea europaea, Prunus dulcis, Vitis vinifera, and Medicago sativa - with different levels of resistance to Xf and found putative genes, pathways and regulatory networks linked to resistance mechanisms.
Materials and methods
Fig. 1. Visual representation of the cross-species transcriptomic analysis pipeline, highlighting the key stages of the workflow. The diagram outlines major components including data preprocessing, reference-based alignment and analysis, de novo transcriptome assembly, and the integration of results for cross-species comparison in response to Xylella fastidiosa infection
A visual summary of the cross-species transcriptomics analysis pipeline showing the major stages of the analysis used in this study is given in Fig. 1. Steps 1 through 5 represent the preprocessing stages of the pipeline conducted before the core RNA-Seq analysis. The pipeline is visually segmented using color codes: the green box highlights the primary steps involved in reference-based analysis, the blue box represents the stages of de novo analysis, and the red box features the main components of the cross-species transcriptomics analysis, including the identification of common and unique differentially expressed genes and subsequent downstream analyses.
Data collection
An exhaustive search was conducted using PubMed and Google Scholar to identify relevant studies on Xf infection. This search was done using species names affected by the bacteria and included articles published up to 2023. Four studies were selected for inclusion in the analysis on the infection dynamics of Xf (Table 1). The selection of the studies was guided by several criteria: (1) their status as economically vital and agriculturally significant crops in regions affected by Xf; (2) documented predisposition to Xf infections based on previous research; and (3) the availability of transcriptomic resources, enabling reliable comparative analyses. These species were chosen to provide a representative framework for studying host responses to Xf across both woody and herbaceous plants. One of the articles examined the responses of different olive cultivars to Xf infection: ‘Leccino’, resistant to the disease [74] and ‘Ogliarola salentina’ highly susceptible, as reported by [31]. In addition to these contrasting olive cultivars, the selected studies explored Xf infection in other plant species, including Prunus dulcis, Vitis vinifera, and Medicago sativa. For olives, we decided to present the data for tolerant and susceptible varieties separately, leading to five comparisons (C1, C2, C3, C4, C5). For further analysis, accession IDs corresponding to the selected studies were collected. The raw sequencing data retrieval was performed using the command-line tool fasterq-dump, which is an integral component of the SRA Toolkit. The raw sequencing data associated with the identified research articles was accessed through the Short Read Archive (SRA) database, curated by the National Center for Biotechnology Information (NCBI) and served as the primary repository.
Table 1. Data sources for cross-species transcriptomic analysis of plant resistance to Xylella fastidiosa infectionComparisonPlantSRA IDRead typeArticleControlTreatedC1Olea europaea cv. LeccinoSRR3310956 SRR3310957SRR3310953 SRR3310954 SRR3310955SingleGiampetruzzi et al. [31]C2Olea europaea cv. Ogliarola salentinaSRR3310961 SRR3310962SRR3310958 SRR3310959 SRR3310960SingleGiampetruzzi et al. [31]C3 Prunus dulcis SRR18264544 SRR18264543 SRR18264542 SRR18264541SRR18264540 SRR18264539 SRR18264538 SRR18264537PairedMoll et al. [71]C4 Medicago sativa SRR10056629 SRR10056630 SRR10056631SRR10056632 SRR10056633 SRR10056634PairedAbou Kubaa et al. [1]C5 Vitis vinifera SRR5851306 SRR5851303 SRR5851304 SRR5851297 SRR5851298SRR5851301 SRR5851302 SRR5851299 SRR5851300 SRR5851305SingleZaini et al. [119]
Data preprocessing, quality checking and read alignment
Raw reads from all five comparisons went through standardized processing in the same preprocessing pipeline to minimize the risk of misinterpretation due to poor data quality or contamination and to ensure consistency across species. Initially, the FastQC tool (v0.12.1) was used to evaluate sequence quality, GC content, presence of adapters, over-represented sequences, and duplication levels. Next, the Cutadapt tool (version 3.7) was applied to remove adapter sequences to ensure that only the actual biological sequences were retained for downstream analysis. For read 1 & 2, adapter trimming was performed in two steps, beginning with a 10-base minimum overlap for the first adapter, followed by the removal of the second adapter with “-b” option. Subsequently, a custom Perl script was executed to remove low-quality reads, retaining only those with a length of ≥ 30 bases, as shorter reads do not significantly contribute to the accuracy of reference genome alignment. Then, to eliminate the misinterpretation due to contamination, the bowtie2 (version 2.5.1) tool was employed for removing the contamination by filtering out reads aligning with known contaminant sequences to remove the unwanted sequences in the dataset. After eliminating rRNA contamination, the pre-processed reads were aligned to their respective reference genomes obtained from the NCBI, except for comparison C4, for which the appropriate reference genome was unavailable. The alignment was conducted using the HISAT2 tool (version 2.2.1) with the default parameters. Alignment indices were built individually for each species’ genome to ensure accurate mapping. Next, the alignment statistics were collected using the flagstat command-line tool, a component within the SAMTools (Version: 1.17) suite. As the datasets were generated using similar sequencing platforms (Illumina) and exhibited comparable quality profiles, no species-specific adjustments were made.
Because of the absence of a suitable reference genome, de novo transcriptome assembly procedure was applied on the pre-processed reads from C4. Trinity (v2.15.1) assembler, De Bruijn graph-based assembler, created the assembly from scratch without relying on any existing genetic blueprint. Following the initial assembly, the CD-Hit (version 4.8.1) clustering tool was applied to remove any redundant sequences among the assembled contigs to streamline the dataset and improve downstream analysis. The quality and completeness of the assembly were assessed with BUSCO (version 5.4.6) which identifies how well the assembly represents a set of conserved single-copy genes that are expected to be present in the genome. Subsequently, TransRate (version 1.0.3) was utilized for reference-free quality assessment of the assembly. Transcriptome assembly quality was further evaluated by mapping RNA-Seq reads back to the Trinity-assembled contigs using the HISAT2 aligner. The percentage of aligned reads was calculated to assess transcript completeness, complementing the BUSCO-based evaluation. The process culminates by combining the annotations for the contigs collected by blasting them against NCBI nr database, UNIPROT and TAIR database.
Differentially expressed gene identification
The read summarization of the RNA-Seq data, for all the comparisons except C4, at gene level was performed with featureCounts (Version 2.0.6) tool. The count matrix was fed to the DESeq2, an R-language statistical pack, for the identification and analysis of differentially expressed genes using a negative binomial generalized linear model. The differential expression estimation of C4 was conducted using RSEM (v1.3.1). The expected-count obtained through the estimation was used to build feature count matrix, which is later fed to the DESeq2 package for downstream analysis. Read counts were normalized by DESeq2, which applies a median-of-ratios method that is robust to differences in sequencing depth and RNA composition. As gene expression comparisons were conducted within species (not across species), species-specific gene length biases did not impact the normalization process. Genes with an adjusted p-value less than 0.05 were regarded as statistically significant. The sequences for the differentially expressed genes were blasted against the Arabidopsis thaliana database to obtain the homolog TAIR IDs. The BLAST results were then categorized based on alignment score and e-value to assess the confidence of homology. “Nearly identical” matches were defined as those with a bit score ≥ 1000 and an e-value of 0, indicating exceptionally high sequence similarity. “Highly similar” matches included those with either a bit score ≥ 1000 and a non-zero e-value, or a score between 500 and 999 with an e-value of 0. These thresholds ensured that only strong and biologically meaningful alignments were prioritized for downstream analysis. Common and unique genes across datasets were subsequently identified using a custom Python script.
Gene ontology analysis
The functional annotation of the gene and gene products were obtained with the help of Gene Ontology (GO), a robust bioinformatics resource which systematically classifies genes and their products across diverse species according to their attributes and functions. For better understanding biological roles, gene and gene products were categorized into specific functional categories such as Biological Process (BP), Cellular Component (CC) and Molecular Function (MF). Biological process involves the execution of biological programs through various molecular activities, molecular function describes specific activities performed by individual gene products, typically at the molecular level while cellular component specifies the spatial locations of gene products. The expression profiling of the common genes was carried out with the DAVID tool [93].
Pathway analysis
The analysis and visualization of extensive genomics and transcriptomics data pertaining to individual genes was carried out using MapMan. This application was utilized to visually represent gene expression data within pre-defined metabolic pathways and functional categories using Arabidopsis thaliana mapping file. MapMan annotated the input data consisted of differentially expressed genes with functional categories (bins) provided by the software. These hierarchical functional categories allowed the exploration of pathways of interest.
Protein–protein interaction network analysis
The complex interactions that underpin cellular processes was revealed through protein-protein interaction (PPI) network analysis, by mapping the physical and functional associations between proteins, resulting in the identification of key proteins that play central roles in maintaining cellular integrity or contributing to disease mechanisms. The analysis result was visualized using a graph where nodes represent proteins and edges indicate interactions. The STRING [100] database was used to build the initial network with a high confidence score > 0.9, which was later imported into Cytoscape (Version 3.10.2) software for proper layout and hub protein identification with using degree centrality as the primary metric provided by the cytoHubba plugin [16]. The cluster analysis was conducted using Molecular Complex Detection (MCODE) plug-in [3]. To find better clusters haircut option was selected with other default settings: degree cut-off at 2, node score cut-off at 0.2, k-core cutoff at 2 and maximum depth set to 100.
Results
Preliminary bioinformatic analysis
In this study around 1096 million raw reads were inputted for pre-processing and approximately 1090 million reads successfully passed the pre-processing stage. The aggregate range of raw reads falls approximately between 105 million to 365 million. After the pre-processing, the number of reads available for the downstream analysis were narrowed down to approximately from 104 million to 364 million (Additional File 1: Sheet 1). The pre-processed reads of Olea europaea cv. Leccino and cv. Ogliarola salentina were aligned to the reference genome of cv. Leccino GWHEUUU00000000.1 obtained from China National Center for Bioinformation GSA (Genome Sequence Archive) database https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA015700. For Prunus dulcis the pre-processed reads were mapped against cv. Texas GCF_902201215.1 genome sourced from NCBI, while Vitis vinifera was aligned against cv. Pinot Noir 40,024 GCF_000003745.3, which was downloaded from NCBI. Due to the unavailability of an appropriate reference genome for Medicago sativa, all the pre-processed reads followed a de novo assembly pipeline. The alignment percentage for different samples of Olea europaea (cv. Leccino) ranges from 86.80 to 88.15%, for Olea europaea (cv. Ogliarola salentina) it ranges from 84.97 to 87.62%, for Prunus dulcis it ranges from 94.26 to 95.01%, and for Vitis vinifera from 91.72 to 93.35%. The total number of raw reads, the total number of reads that successfully passed the pre-processing steps, the alignment percentage for individual samples, and the average alignment percentage of each study are available in Additional File 1: Sheet 1 & 2.
For comparison C4, Trinitty assembler produced 302,564 contigs from the pre-processed reads. CD-HIT clustering tool was later applied to reduce the redundancy, and it created 239,531 non-redundant clusters with 90% similarity threshold. The assembly statistics were collected with TransRate tool and is available in Additional File 2: Sheet 1. The completeness of the assembly was assessed using BUSCO and the assessment was carried out with ‘viridiplantae_odb10’ dataset (Additional File 2: Sheet 2). The RNA-Seq reads were mapped back to the Trinity-assembled contigs to cross-verify the assembly quality (Additional File 2: Sheet 3). The annotation for the clustered contigs were gathered from NCBI nr (24454 entries) database, UNIPROT (26234 entries) and TAIR (19450 entries) database and is provided in Additional File 3. Only contigs matching ‘Viridiplantae’ or labeled as ‘unannotated’ were retained for the downstream analysis. The count matrix generated using RSEM was used for differentially expressed gene identification.
Table 2. Summary of differentially expressed genes (DEGs) across five comparisons (p-value < = 0.05 and|log2(fold change)| >0.50)PlantComparisonlog2FC range (min-max)Adjusted p-value cut-off (0.05)Total DEGsUp-regulatedDown-regulatedO. europaea (cv. Leccino)C1(−11.058304) - (08.280662)866441314533O. europaea (cv. Ogliarola salentina)C2(−10.166598) - (21.457315)713632343902 Prunus dulcis C3(−03.992666) - (03.724467)1101550551 Medicago sativa C4(−14.646919) - (15.473057)10,32647025624 Vitis vinifera C5(−09.312933) - (11.010041)658235982984
Differentially expressed genes in response to Xf infection
The differentially expressed genes (DEGs) were analyzed to uncover the genes whose expression levels vary between biological conditions or experimental treatments. A|log₂FC| >0.50 threshold was selected to balance sensitivity with biological interpretability in this cross-species context, where subtle yet conserved expression changes are often biologically meaningful. A stricter cutoff|log₂FC| >1 excluded many conserved genes, potentially overlooking important regulatory signals that are modest in magnitude but evolutionarily conserved and functionally relevant. The results of this sensitivity analysis are shown using an UpSet plot in Additional File 4, demonstrating that a higher cutoff would exclude many conserved expressions. This supports the decision to use a lower threshold for log2fold change. The estimation of differentially expressed genes, aimed at the identification of genes, with p-adjusted value threshold of 0.05 resulted in the detection of a total of 8664 DEGs in Olea europaea (cv. Leccino) among which 4131 were up-regulated and 4533 were down-regulated. In Olea europaea (cv. Ogliarola salentina) the comparison revealed 3234 up-regulated and 3902 down-regulated genes aggregated to 7136 DEGs. 1101 and 6582 were the total number of differently regulated genes found in Prunus dulcis and Vitis vinifera, respectively. Finally, the analysis discovered 4702 up-regulated and 5624 down-regulated genes in Medicago sativa (Table 2). The list of differentially expressed genes is available in Additional file 5. The normalized count for all the genes obtained during the analysis performed with DESeq2 package is available in Additional file 6_1 and in Additional file 6_2.
The variability of gene expression within a dataset is depicted in the dispersion plot (Additional file 7) generated by DESeq2, which assesses the reliability of the differential expression analysis results. In the dispersion plots of O. europaea cv. Leccino, cv. Ogliarola salentina and Prunus dulcis, most of the genes are tightly clustered around the trend line, indicating that the analysis effectively modelled the variability. In Medicago sativa a higher variability of genes is shown, as in Vitis vinifera, the genes are loosely clustered near the trendline.
MA plot in Additional file 8 with alpha threshold of 0.05 depicts the log2fold changes associated with a specific variable with respect to the average expression strength of the genes in the treatment ‘control vs treated’. The plots were generated after applying ‘apeglm’ shrinkage algorithm [122] to remove the noise generated with log2fold changes from low count genes. The differentially expressed genes are represented with blue dots, whereas grey dots represent the non-significant genes. A symmetrical distribution of points around the horizontal axis of the plot is visible in the initial four studies which indicates a lack of systematic biases whereas, in comparison five, a more scattered distribution is found.
Gene identification was performed through cross-study comparisons using BLAST alignment against the TAIR database, retaining only entries with “Highly similar” or “Nearly identical” similarity labels for TAIR ID conversion. The analysis revealed 18 common differentially expressed genes (DEGs) across all five studies, visualized in the overlapping quintuple intersection of Fig. 2’s Venn diagram and the details about these genes are available in Table 3. Species-specific unique DEGs varied substantially: Olea europaea cv. Leccino exhibited 495 unique DEGs, contrasted with 186 in cv. Ogliarola salentina. Prunus dulcis contained 321 unique DEGs, Medicago sativa 189, and Vitis vinifera showed the highest divergence with 661 unique DEGs. The genomic architecture of resistance has not been fully explored and clarified in V. vinifera, which could explain the significant number of unique differentially expressed genes (DEGs) identified in this species. All domesticated grapevines are susceptible to Pierce disease, suggesting that the genetic basis for resistance is lacking in V. vinifera [73]. This lack of resistance may lead to a higher expression of stress-related genes in response to infection, resulting in the identification of 661 unique DEGs in this cultivar. Environmental factors may also influence the expression of DEGs in V. vinifera, potentially exacerbating its susceptibility to Xylella fastidiosa. The findings imply that the unique DEGs in V. vinifera could be a response to the pathogen’s presence, reflecting the plant’s inability to mount an effective defense due to the absence of resistance genes found instead in its wild relatives. All common DEG expression profiles, TAIR annotations, and comparison-specific unique gene sets are systematically catalogued in Additional File 9.
Fig. 2. Venn diagram showing the overlapping and unique differentially expressed genes (DEGs) identified across five comparisons. The overlapping section highlights common Differentially Expressed Genes (DEGs), while the individual circles represent unique DEGs for each comparison. C1 corresponds to Olea europaea cv. Leccino, C2 to Olea europaea cv. Ogliarola Salentina, C3 to Prunus dulcis, C4 to Medicago sativa, and C5 to Vitis vinifera
Table 3. Common genes consistently expressed in response to Xylella fastidiosa across five comparative analysesGene nameTAIR IDProtein nameFunctional category TPS9 AT1G23870Trehalose 6-phosphate synthaseResource allocation and transport. SKS5 AT1G76160SKU5 similar 5Structural defense and cell wall remodelling. BAS AT1G78950Beta-amyrin synthaseSecondary metabolism and antimicrobial effectors. SBT1.8 AT2G05920Subtilase family proteinSecondary metabolism and antimicrobial effectors. KCS11 AT2G266403-ketoacyl-CoA synthase 11Lipid metabolism and membrane integrity. PED1 AT2G33150Peroxisomal 3-ketoacyl-CoA thiolase 3Lipid metabolism and membrane integrity. PDR6 AT2G36380Pleiotropic drug resistance 6Secondary metabolism and antimicrobial effectors PIC30 AT2G39210Major facilitator superfamily proteinStructural defense and cell wall remodelling. OFUT20 AT2G44500O-fucosyltransferase family proteinStress signaling and hormonal crosstalk. CYP707A4 AT3G19270Cytochrome P450 707A4Stress signaling and hormonal crosstalk. DTX40 AT3G21690MATE efflux family proteinCalcium signaling and circadian regulation. ACA12 AT3G63380ATPase E1-E2 type family proteinCalcium signaling and circadian regulation. VEP1 AT4G24220VEIN PATTERNING 1Structural defense and cell wall remodelling. ACA2 AT4G37640Calcium-transporting ATPase 2Calcium signaling and circadian regulation. SDP1 AT5G04040SUGAR-DEPENDENT 1Resource allocation and transport. AAP2 AT5G09220Amino acid permease 2Resource allocation and transport. AOS AT5G42650Allene oxide synthaseStress signaling and hormonal crosstalk. KAS1 AT5G462903-ketoacyl-acyl carrier protein synthase ILipid metabolism and membrane integrity.
Gene ontology analysis of Xf associated DEGs
Gene ontology (GO) enrichment analysis of differentially expressed genes (DEGs) identified 631 significantly up-regulated and 686 down-regulated terms (p < 0.05). Biological processes (BP) showed comparable enrichment between directions, with 345 positively and 362 negatively regulated terms (Fig. 3). Cellular component (CC) terms followed this pattern (96 up-regulated, 106 down-regulated), while molecular function (MF) terms exhibited stronger down-regulation (218 vs. 190 up-regulated). Notably, 5% of up-regulated genes mapped to ‘defense response to bacterium’ (BP). CC analysis revealed bidirectional enrichment for chloroplast, cytoplasm, and membrane localization. In MF, protein/ATP/metal ion binding dominated down-regulation, while cytoplasmic and organelle-related terms emerged in up-regulated MF annotations (Additional File 10).
Fig. 3. Summary of the top twenty Gene Ontology (GO) terms identified through GO enrichment analysis of differentially expressed genes (DEGs) obtained from five comparisons. Only terms with a p-value less than 0.05 are regarded as enriched. (A) Up-regulated BP (Biological Process) terms (B) Down-regulated BP terms. (C) Up-regulated CC (Cellular Component) terms (D) Down-regulated CC terms (E) Up-regulated MF (Molecular Function) terms (F) Down-regulated MF terms
Pathway analysis
MapMan analysis revealed substantial Xf-induced modulation of biotic stress pathways, with 463 differentially expressed genes (DEGs) identified in the ‘Biotic Stress’ pathway (Fig. 4). The transcriptional scenario showed 249 up-regulated genes (potentially encoding defense-related activators) and 209 down-regulated genes (indicating pathway suppression), alongside five constitutively expressed genes conserved across all comparisons - putative core regulators of biotic stress adaptation. Prominent examples include the up-regulation of the PIN1 and PIN6, which enhances auxin-responsive gene expression [83, 114], and the down-regulation of the genes TF ARF7, which disrupt auxin transport, adversely affecting growth and defense [108]. In the brassinosteroid pathway, the gene CYP85A2 was down-regulated [46] altering plant immunity through its interactions with other pathways [26, 53], while BAS emerged as a key modulator of brassinosteroid signaling, of abscisic acid (ABA) metabolism, a coordinated modulation of ABA-related signaling and growth regulatory pathways was observed where ABA1 was down-regulated and GCR2, an ABA receptor, was up-regulated, potentially indicating a feedback mechanism or enhanced ABA sensitivity [22, 60]. The upregulation of ETR2 during bacterial infection may reflect a feedback mechanism in response to elevated ethylene levels or ethylene-induced stress conditions, as ethylene receptors are often transcriptionally induced by ethylene itself [8], while the jasmonate pathway, involving AOS and LOX1, was critical for jasmonic acid production, which fortifies defense mechanisms [18, 64, 65, 109]. Moreover, the down-regulation of ATCSLD5 shows a strategic shift in cell wall metabolism during pathogen assaults [7], and XTH6 overexpression enhanced resistance through increased cell wall remodelling [36]. MAPK signaling networks were also implicated in plant immunity [50].
Fig. 4. MapMan-generated general overview pathway of common and unique differentially expressed genes (DEGs) illustrating the impact of Xylella fastidiosa infection on the ‘Biotic Stress’ pathway, highlighting the differentially regulated genes in response to biotic stress conditions
Fig. 5A) MapMan overview of metabolic activity related to shared and specific differentially expressed genes (DEGs) summarizing the overall gene expression changes in response to Xylella fastidiosa (Xf) infection. B) The proteasome pathway enriched among shared and distinct DEGs identified in response to Xf infection
Parallel metabolic profiling through the ‘Metabolism Overview’ pathway (Fig. 5 A) identified 376 DEGs, maintaining the five conserved genes while revealing distinct regulatory patterns: 153 significantly up-regulated versus 218 down-regulated loci. This bidirectional modulation highlights Xf’s dual strategy of hijacking metabolic resources while triggering counter-defense mechanisms. The list of genes along with the bin labels and descriptions are available in Additional File 11.
Proteasome profiling through the ‘Ubiquitin Dependent Degradation’ pathway (Fig. 5B) identified 48 DEGs, revealing distinct regulatory patterns: 12 significantly up-regulated versus 36 down-regulated loci. This bidirectional modulation highlights a possible Xf’s dual strategy of hijacking ubiquitin-proteasome system resources while triggering counter-defense mechanisms.
PPI network construction and analysis
Fig. 6A) Protein-protein interaction (PPI) network of defense-related proteins, highlighting the 10 most significant hub proteins identified within the network. B) PPI network of common and unique differentially expressed genes (DEGs), showcasing the 10 leading hub proteins and their interactions. C) The three most relevant clusters derived from the defense related PPI network through network cluster analysis
Protein-protein interaction networks were reconstructed using STRING-derived associations (confidence score > 0.9) from differentially expressed genes, followed by Cytoscape visualization and topological analysis. Network complexity was reduced through two complementary strategies:
- Hub prioritization: CytoHubba identified top 10 hub nodes per network via degree centrality (direct connection count), visualized as inner-circle core proteins versus outer-circle peripheral nodes.
- Pathway pruning: First-neighbor interactions of hub proteins were retained to emphasize primary signaling axes.
The newly constructed network only shows the first-stage nodes displaying the shortest path. In the PPI networks, the inner circle shows the hub proteins, whereas the outer circle shows the other proteins.
The defense-specific PPI network (Fig. 6 A, confidence > 0.4) revealed 10 highly interactive proteins - WRKY33, MPK6, BAK1, HEL, NPR3, RAR1, EIN3, WRKY40, CERK1 and FMO1 - where WRKY33, MPK6, and BAK1 as the top interactors among 10 critical hubs - all established immune signaling components.
Contrastingly, the pan-DEG network (Fig. 6B, degree cutoff ≥ 15) identified PGIC (plastidial glucose transporter) and CSY4 (citrate synthase) as central metabolic hubs, alongside tandem kinases TKL-1/TKL-2 (transketolase-like proteins) and starch metabolism regulators (MLS, PGMP). This topology suggests a two-pronged adaptation to Xf infection:
- Resource reallocation: Upregulated carbon metabolism (GGAT2, SDP1) and lipid remodelling enzymes.
- Signaling rewiring: High-degree interactors T16L4.190 (uncharacterized transporter) and T22P22.110 (ABC transporter-like).
MCODE clustering revealed three functional modules, with BAK1 (LRR receptor kinase) serving as the seed node of Cluster 1 (Fig. 6 C). This defense-signaling nexus contained pathogen-responsive receptor kinases (e.g., CERK1, BAK1) and PR protein interactors (thaumatin-like proteins, β−1,3-glucanases). Full network annotations– including kinase domains (TKL-1/2; IPR017441), metabolic domains (PGIC; PF00359), and cluster-specific interactomes– are detailed in Additional File 12.
The species-specific PPI networks in Additional File 13 depict the core hub proteins in each species. These species-specific interactions may underlie the differences in how each host responds to Xylella fastidiosa infection, potentially influencing the severity of symptoms, effectiveness of pathogen restriction, and overall susceptibility or tolerance. Each network highlights the top 10 hub proteins. The hub proteins—including T16L4.190, T22P22.110, USP, MLS, and CSY4—are shared between comparisons C1 and C2, while unique proteins in each network highlight their differing resistance to Xf. The presence of SDP1 as a common protein in the species-specific networks constructed for C2 and C4 suggests a shared metabolic adaptation strategy, where both species rely on sugar metabolism to support their defense mechanisms. The presence of PGMP, a starch metabolism regulator, in both C4 and C5 indicates the commonality points to functional convergence in their response to bacterial pathogens.
Discussion
We identified a novel, conserved core set of 18 genes shared across four phylogenetically diverse species (Olea europaea, Prunus dulcis, Vitis vinifera, and Medicago sativa), suggesting the existence of a fundamental and ancient molecular module underpinning resistance to Xylella fastidiosa. These genes represent key players in stress response, signaling, and defense mechanisms essential for plant resilience. Their roles are highlighted below, drawing from their known functions in other species and in various stress conditions:
Structural defense and cell wall remodelling (Common genes)
SKS5 (AT1G76160), a multicopper oxidase gene is associated with the regulation of the apoplast fluid proteome during systemic acquired resistance (SAR) [12, 39]. SKU5 has been implicated in modulating plant responses to bacterial pathogens, including Pseudomonas syringae [45]. Its role in enhancing pathogen resistance suggests its potential contribution to Xf resistance through proteome modification in the host’s extracellular space.
The gene PIC30 (AT2G39210) is involved in the regulation of plant cell-wall composition and responses to biotrophic pathogens [94]. Its role in modulating the plant’s response to cell wall damage suggests a possible function in enhancing the plant’s resistance to Xf by fortifying physical barriers against pathogen invasion. VEIN PATTERNING 1 (AT4G24220), primarily associated with vein architecture, has also been linked to defense responses [40, 102]. Its upregulation during Xf invasion may play a role in preserving vascular integrity.
Stress signaling and hormonal crosstalk (Common genes)
The O-fucosyltransferase (AT2G44500) OFUT20 or F4I1.31 gene is associated with hormone signaling, particularly in response to pathogen infections [77, 118]. Hormones like jasmonic acid (JA) and salicylic acid (SA) are crucial for systemic defense responses [106]. Its involvement in Xf resistance may relate to modulation of these signaling pathways, improving the plant’s immune function. The Allene oxide synthase (AOS) - AT5G42650 - is involved in the biosynthesis of jasmonic acid, a hormone critical for the plant’s defense against necrotrophic pathogens [29, 97]. AOS’s role in enhancing jasmonate production suggests it may be integral to the plant’s ability to respond effectively to Xf. CYP707A4 (AT3G19270) is a cytochrome P450 enzyme that plays a key role in the catabolism of abscisic acid (ABA), a hormone involved in stress responses. CYP707A4 helps regulate ABA levels, which in turn influences the plant’s resistance to osmotic stress [15] and possible pathogens. Its activity may enhance Xf resistance by modulating the plant’s stress response pathways.
Lipid metabolism and membrane integrity (Common genes)
The KCS11 gene (AT2G26640) encodes a ketoacyl-CoA synthase, which is crucial in the synthesis of fatty acids and waxes, contributing to the plant’s resistance to abiotic and biotic stresses, including cold and salt stress [62]. Given that Xf infection triggers oxidative stress, KCS11’s role in maintaining cell membrane integrity and lipids could help mitigate damage and support resistance. The gene PED1 or KAT2 (AT2G33150) is a gene encoding a thiolase with demonstrated importance in β-oxidation and lipid metabolism [30]. This suggests a potential role in mitigating the oxidative burst triggered by Xf infection. The gene KAS1 (AT5G46290) is involved in fatty acid biosynthesis, and according to TAIR, it can have a crucial role in plant defense mechanisms. The production of fatty acids during pathogen stress can influence membrane integrity and synthesis of defense molecules.
Secondary metabolism and antimicrobial effectors (Common genes)
The BETA-AMYRIN SYNTHASE (BAS - AT1G78950) gene is crucial for the formation of triterpenoid compounds, which play an essential role in plant defense against pathogens and may be involved in SAR [39]. In this context, this gene might contribute to the synthesis of protective compounds, enhancing defense mechanisms against Xf in woody species like olive and grapevine. The PDR6 or ABCG34 (AT2G36380) is a member of the ABC transporter family and has been shown to mediate the efflux of the defense compound camalexin in Arabidopsis [68]. By enhancing the removal of toxins or secondary metabolites produced during Xf infection, PDR6 likely supports the plant’s ability to resist infection. Plant immune signaling largely relies on post-translational modifications to establish a rapid and appropriate defense response to different pathogen types and infection pressure. [68] demonstrated that PDR6 plays a major role in transporting camalexin, as indicated by the significantly lower camalexin secretion in PDR6 mutants compared to wild-type plants. The phosphorylation of PDR6 in Arabidopsis by two AGC kinases, OXI1 and AGC2-2, led to increased camalexin levels on leaf surfaces and decreased levels in the leaf interior, enhancing resistance to pathogens such as the Gram-negative bacterium Pseudomonas syringae and the fungus Botrytis cinerea. To date, no QTLs have been mapped specifically for resistance to Xylella fastidiosa in olive. However, BAS and PDR6 genes were identified in Giampetruzzi (2016) ’s RNA-seq study, and the same genes were also detected in Leccino progenies by [51]. Notably, in La Notte’s study (2024), resistant seedlings S215 and S234 appeared to escape infection 24 months post-inoculation, with most of their progenies—especially those of S215—showing recovery. BAS and RD6 were among the DEGs common to both genotypes. SUBTILASE 1.8 (AT2G05920) genes are serine proteases that have been shown to regulate plant immune responses by modulating the activity of other defense-related proteins [39]. Subtilase genes could be involved in processing proteins that activate immune pathways in response to Xf infection.
Resource allocation and transport (Common genes)
The TREHALOSE-6-PHOSPHATASE SYNTHASE 9 (AT1G23870) gene is involved in the biosynthesis of trehalose, a sugar known for its protective role under stress conditions [88]. In Arabidopsis, trehalose accumulation has been linked to enhanced tolerance against osmotic stress, bacterial infections, and heat stress (reviewed by [21]. Given the environmental stressors associated with Xf infection, AT1G23870 may help in maintaining cellular integrity during pathogen-induced stress. The gene SDP1 (AT5G04040) is involved in sugar metabolism, which is essential for energy production during stress responses. In the case of Xf infection, SDP1 could support the energy requirements needed for defensive responses, such as the synthesis of secondary metabolites and reactive oxygen species (ROS). The AAP2 (AT5G09220) is an amino acid permease that regulates amino acid transport, which is critical for plant immune responses [14]. During Xf infection, AAP2 may help facilitate the uptake of essential amino acids needed for the synthesis of defense-related proteins and metabolites.
Calcium signaling and circadian regulation (Common genes)
ACA2 (AT4G37640) and ACA12 (AT3G63380) are calcium-transporting ATPases that play distinct roles in calcium signaling during plant immune responses, differing in their localization, regulatory mechanisms, and functions. ACA12 is situated at the plasma membrane and operates independently of calmodulin, enabling rapid calcium efflux that supports early immune responses such as pathogen-triggered immunity (PTI) and stomatal closure, which are essential for blocking pathogen entry. In contrast, ACA2 which is localized to the endoplasmic reticulum, where it is not directly activated upon pathogen perception, maintains the internal Ca²⁺ balance required for proper functioning of downstream signaling pathways, including those controlling gene expression and cell survival [59, 79, 117]. The MATE efflux family protein (AT3G21690) gene is involved in aluminium stress [95] and circadian rhythm regulation, which has been shown to influence plant immune responses. Plants with well-regulated circadian rhythms are more effective in defense responses against pathogens [42]. Its role in Xf resistance may be connected to the timing of immune activation.
The conserved genes illustrate a comprehensive strategy against Xf, consisting of several layers:
First, pre-invasive defenses such as cuticular waxes (KCS11, KAS1) and possible cell wall reinforcement (PIC30) to block pathogen entry. Next, stress signaling mechanisms, including hormonal crosstalk (AOS, CYP707A4) and calcium signaling (ACA12), amplify immune responses. Following invasion, post-invasive responses deploy antimicrobial compounds (BAS, PDR6) and proteolytic activation (SUBTILASE 1.8) to target Xf proliferation. Finally, resource optimization through trehalose (AT1G23870) and amino acid transport (AAP2) ensures metabolic demands are met during infection.
Biotic stress pathway
The biotic stress pathway elucidates how plants regulate defenses against Xf biotic stressors through hormone interactions. Salicylic acid induces calcium ion (Ca²⁺) bursts by activating cytosolic Ca²⁺ channels, which subsequently amplify immune responses through the activation of pathogenesis-related (PR) genes. However, Xf can subvert this process by degrading SA or inhibiting PR gene expression. Jasmonic acid promotes anti-herbivore responses, thereby prioritizing these defenses over SA-mediated immunity. It activates ACA2, a plasma membrane Ca²⁺-ATPase, which exports cytosolic Ca²⁺, thereby diminishing the synergy between SA and calcium signaling. This antagonism may be exploited by Xf to weaken plant defenses further, as the regulation of calcium homeostasis is crucial during stress responses [28, 34, 75]. Abscisic acid activates ACA12 under drought stress, sequestering Ca²⁺ into the endoplasmic reticulum and reducing cytosolic Ca²⁺ levels. Xf infection is perceived as a condition of drought stress, elevating ABA levels. This can consequently impact calcium signaling pathways that are vital for plant immunity [13]. In the early stages of infection, SA predominates, while in chronic stages, ABA and JA become dominant, leading to suppression of defense mechanisms. This shift in hormonal balance reflects the complex regulatory networks that govern plant responses to pathogens, highlighting the need for further exploration of these interactions [55, 119]. For instance, it would be crucial to check how hormonal levels change and modulate during the infection. Auxins play their part by balancing growth and defense through PIN1 and PIN6 genes [83, 114]. Brassinosteroids enhance defenses by activating stress-related genes [46], while ethylene and jasmonate pathways synergize to combat Xf (AOS/LOX1; [18, 65]. Cell wall integrity is affected by ATCSLD5 gene [7] but reinforced by XTH6 gene [36]. Immune regulation involves MLO genes, MAPK signaling [50], and the ubiquitin-proteasome system [121], with BAS further enhancing immunity through brassinosteroid signaling crosstalk [26]. These insights into molecular mechanisms provide valuable targets for improving crop resilience and promoting sustainable agriculture.
Metabolic overview
MapMan’s metabolic overview offers a visual representation of how bacterial infections influence plant metabolism, showing the significant alterations in metabolic pathways that occur upon infection. The plant cuticle is a lipid-based polymer layer coated with waxes that covers the outermost surfaces of plant organs, acting as the first line of structural defense against pathogens. Instead of remaining static, the cuticle undergoes dynamic remodelling during pathogen invasion and is involved in plant immune signaling. The common genes involved in various metabolic pathways include KAS and KCS in lipid metabolism, ATTPS9 in carbohydrate metabolism, PKT3 in amino acid metabolism, and BAS in secondary metabolism.
The β-Ketoacyl-ACP Synthase (KAS) genes are integral to the fatty acid biosynthesis pathway in plants [58], producing fatty acids essential for constructing lipid-based structures like cell membranes and the cuticular layer. These structures serve as primary physical barriers against pathogenic invasions, including bacterial infections. Fatty acids synthesized through the KAS pathway are precursors for signaling molecules such as jasmonic acid (JA), which play pivotal roles in activating plant defense mechanisms. The KCS gene family plays a key role in producing very-long-chain fatty acids (VLCFAs), essential for forming protective plant barriers like cuticular waxes and suberin. These barriers help resist pathogens. Research shows some KCS genes are activated during infection while others are suppressed, indicating a gene-specific regulatory system that shapes plant defense responses (e.g. [82].
The ATTPS9 gene, classified under the minor CHO (carbohydrate) section, encodes an enzyme involved in trehalose-6-phosphate synthase activity, which is crucial for stress responses and energy regulation in plants [107]. The PKT3 gene, associated with amino acid metabolism, encodes an enzyme involved in polyketide biosynthesis—secondary metabolite that influence plant-pathogen interactions—while polyketide synthase (PKS) pathways, including the PKS10-2 pathway, are crucial for the pathogenicity of certain plant pathogens and may play roles in defense mechanisms against bacterial infections in host plants (e.g. [103]. The β-Amyrin Synthase (BAS) contributes significantly to the plant secondary metabolism by catalyzing the production of β-amyrin, which can induce the production of ROS, suggesting its potential as an antimicrobial agent [39].
Ubiquitin dependent degradation
In plants, protein degradation is tightly regulated by the ubiquitin–proteasome system (UPS), which serves as a core component of immune and stress responses. In addition to its proteolytic role, the UPS—through its 20 S RNase activity—may participate in a still unidentified antiviral defense mechanism by modifying the proteome to enhance survival. Variations in ubiquitin levels, as well as in the activity of E1 and E2 enzymes, can lead to widespread cellular reprogramming during defense, while E3 ubiquitin ligases, which determine substrate specificity, are critical players in plant–pathogen interactions [20]. The up-regulation of UBQ4, a polyubiquitin gene, during pathogen infection indicates an increased demand for ubiquitin monomers to support the ubiquitin–proteasome system (UPS), which is essential for tagging and degrading misfolded or regulatory proteins, allowing the plant to fine-tune its immune response [99]. The expression of the E1-activating enzyme gene SAE2 and the SUMO‑conjugating E2 enzyme UBC9 enhances post-translational modifications critical for immunity [10]. Zinc finger E3 ligases facilitate selective protein ubiquitination, modulating immune receptor levels, signaling intermediates, and transcriptional regulators essential for both PTI and ETI pathways [115]. F-box proteins dynamically regulate target recognition for SCF complexes, impacting defense signaling and strengthens immunity [98], whereas degradation of key F-box proteins like COI1 [35] hinders jasmonate signaling and compromises immune responses. The marked induction of proteasome subunit expression shows that, during basal defense, the plant boosts proteasome activity to rapidly degrade specific proteins, helping to regulate and fine-tune immune signaling, remove harmful or unnecessary proteins, and thereby strengthen its ability to resist infection [105]. However, to date, there is no evidence that Xylella fastidiosa interferes with proteasome activity to disrupt phytohormone signaling; therefore, further studies should investigate whether Xf effectors can interact with the UPS. This could be achieved through effector screening approaches, such as yeast two-hybrid assays or affinity purification–mass spectrometry, to identify potential interactions between Xf secreted proteins and host UPS components.
Hub proteins
The protein-protein interaction (PPI) analysis revealed a complex network of genes involved in the response to Xylella fastidiosa infection. By mapping these interactions several highly interconnected hub nodes emerged which suggested the presence of functional pathways and modules related to resistance. In particular, the analysis of interactions between defense-related proteins highlighted several proteins known for their involvement in immune responses, such as BAK1 (cluster 1 Fig. 6 A, C), RIPK (cluster 2, Fig. 6 C) and MAKSA (cluster 3 Fig. 6 C). The network-driven identification of BAK1 as a dual topological and functional hub, alongside metabolic regulators (PGIC, CSY4), reveals a sophisticated interplay between resource management and immune prioritization in Xf-infected plants. These findings align with the emerging paradigm of “defense priming through metabolic reconfiguration”, where plants optimize resource allocation to sustain energetically costly immune responses. The prominence of receptor kinases (BAK1, CERK1) and PR protein interactors in Cluster 1 underscores the activation of canonical pathogen recognition mechanisms. Its interactions with MPK6, WRKY33 and WRKY40 suggest BAK1’s central role in defense signaling [112]. WRKY33 and WRKY40 are significant in modulating immune responses, with WRKY33 linked to oxidative stress and jasmonic acid signaling, while WRKY40 enhances resistance against pathogens and abiotic stress [113].
The hub protein BAK1, also known as SERK3, plays a crucial role in plant immunity, including potential resistance to Xanthomonas fastidiosa and Xylella. One key characteristic of BAK1 is its function as a coreceptor for multiple receptor-like kinases (RLKs), which are essential for recognizing pathogen-associated molecular patterns (PAMPs) and initiating immune responses. BAK1 interacts with pattern recognition receptors (PRRs) such as FLAGELLIN-SENSING2 (FLS2) and ELONGATION FACTOR-TU RECEPTOR (EFR), facilitating the activation of immune signaling pathways upon pathogen detection [17]. Another important feature of BAK1 is its proteolytic processing, which is regulated by calcium and is critical for its function in plant immunity. The cleavage of BAK1 enhances its ability to mediate immune responses, as evidenced by the identification of the aspartic acid residue at position 287 (D287) as essential for this process. Mutations at this site, such as BAK1D287A, impair the protein’s ability to activate downstream signaling pathways, leading to compromised immune responses. BAK1’s localization to the plasma membrane is vital for its role in immunity. Proper localization allows BAK1 to interact with other RLKs and initiate signaling cascades that lead to defense responses against pathogens like Xf. The D287 mutation not only affects BAK1’s cleavage but also reduces its plasma membrane localization, further highlighting the importance of this residue in maintaining BAK1’s functional integrity in plant defense against Xanthomonas and Xylella in transgenic sweet orange and Arabidopsis [32, 69]. Notably, [51] also reported BAK1 among the DEGs in the progenies of resistant genotypes S234 and S215, further supporting its potential role as a hub gene in the defense response against Xylella fastidiosa.
MPK6, a mitogen-activated protein kinase, integrates defense signals and activates downstream responses, including ROS production [111]. Proteins like NPR3 and EIN3 are part of hormonal signaling networks that mediate defense responses, with NPR3 regulating SAR [25] and EIN3 involved in ethylene signaling against pathogens [120]. CERK1, known for chitin perception, may indicate broader defense mechanisms against Xf. ALD1 is another interesting node identified in the network, having a relationship with BAK1. In model plants, it has been demonstrated that ALD1 plays a crucial role in establishing robust defense responses against pathogens. The ALD1 mutant is shown to be hypersusceptible to the virulent bacterial pathogen Pseudomonas syringae, which correlates with a reduced accumulation of the defense signal salicylic acid (SA). This suggests that ALD1 is vital for effective immune responses, particularly in the context of pathogen resistance [96].
Another important node is represented by HEL (Hevein-like) genes, crucial in plant defense responses to biotic stress, particularly through their interaction with various hormones [2]. Reactive electrophile species, which accumulate during pathogenesis, can activate HEL gene expression, indicating their role in the plant immune response. Specifically, the expression of HEL is upregulated by exogenous OPDA (oxo-phytodienoic acid), a compound that is part of the jasmonate family, highlighting the connection between HEL genes and jasmonate signaling [2]. Moreover, the study suggests that HEL gene expression is also influenced by salicylic acid and other low-molecular-mass regulators, which are known to control defense gene expression in plants. This interplay between HEL genes and signaling molecules like jasmonate and salicylic acid underscores the complexity of the plant defense mechanisms against biotic stress [2].
The initial events associated with pathogen perception and responses to environmental stresses encompass the generation of ROS (e.g. [70] and nitric oxide (review in [92], both of which are integral to the disease process, although their precise roles remain the subject of ongoing investigation. Along with ROS and reactive nitrogen species, reactive electrophile species (RES) play key role in regulating defense genes. They may also cause damage to both host and pathogen cells, which is a common aspect of the plant’s response to non-virulent pathogens [2].
Since it is well known that Xylella fastidiosa disrupts phytohormone networks to promote infection [5, 81], future investigations should explore whether Xf can interfere, for example, with HEL expression by modulating RES and ROS, potentially blocking the production of hevein-like proteins. This hypothesis is supported by the findings that suggest Xf employs various extracellular proteins to manage oxidative stress and facilitate biofilm formation, which is crucial for its pathogenicity [67].
In Cluster 2, Fig. 6 C, the protein RIPK is crucial for the production of ROS, which are key signaling molecules in plant defense mechanisms. These mechanisms operate across various layers of the plant immune system, including effector-triggered immunity (ETI), pathogen-associated molecular pattern-triggered immunity (PTI), damage-associated molecular pattern-triggered immunity (DTI) and SAR. Model plants with mutations in the RIPK gene produce lower levels of ROS when exposed to immune elicitors, indicating that RIPK is essential for ROS generation and consequently, for effective plant defense [56]. HSP90.1 and HSP90.2 are closely related members of the HSP90 (Heat Shock Protein 90) family in plants. As a molecular chaperone, HSP90-1 ensures the proper folding, stability and activation of various client proteins involved in defense mechanisms. It interacts with RAR1 and SGT1 to regulate the stability and function of Resistance (R) proteins, which are crucial for triggering defense signaling pathways [101]. HSP90.2 acts as a partial suppressor of the autoimmune mutant chs3-2D in Arabidopsis thaliana and thus it is essential for CHS3-mediated defense signaling [61]. SRFR1 plays a key role in modulating the balance between defense activation and suppression by acting as a negative regulator of effector-triggered immunity against the bacterial pathogen and thereby fine-tuning the immune system to balance defense and growth [57].
In Cluster 3, MAC5A plays a role in mRNA splicing during pathogen defense, emphasizing its relevance in gene expression regulation [72, 110].
The observed metabolic alterations among the four species, marked by the upregulation of genes associated with carbon metabolism (e.g., GGAT2, SDP1) and the regulation of ABC transporters (T22P22.110), suggest two plausible compensatory strategies employed by the plant. The first, nutrient deprivation defense, involves redirecting photoassimilates from the xylem to parenchyma cells via PGIC, to starve the vascular pathogen. The second strategy, toxin compartmentalization, suggests that ABC transporters may play a role in sequestering Xf-derived toxins. This has also been reported in Citrus species [23], where intriguingly, Xf in vitro counteracts the plant defense responses through osmoregulated periplasmic glucans (OPGs) synthesized by MDOH (XF1623) and MDOG (XF2682)2. These OPGs stabilize membrane integrity under nutrient stress—a direct adaptation to survive the host’s xylem-specific starvation tactics, thriving under stress conditions within the xylem of its host plants and inducing acid tolerance [23]. Such co-evolutionary dynamics are further exemplified by the redox rewiring response, which is also observed in the four species making it possible to counteract the ROS-neutralizing catalases produced by Xf, as discussed by [31] and [51] further illustrating the intricate interplay between the pathogen and host defense mechanisms.
The frequent appearance of BAK1 across different species’ networks suggests that these plants may depend on BAK1-driven PTI as a core component of their defense system. Its interaction with BIR1—a key suppressor of cell death and immune responses—further highlights the existence of a tightly regulated mechanism that prevents unwarranted immune activation [63]. The overlap of BAK1 and BIR1 suggests that despite evolutionary divergence, these species may maintain similar defense signaling modules, possibly indicating shared resistance strategies to biotic stressors. Similarly, the presence of RIPK (RPM1-Induced Protein Kinase) across majority of species-specific PPI networks suggests it’s functions as a conserved immune regulator, particularly through ROS-mediated defense signaling [56]. RIPK activates the NADPH oxidase RBOHD, promoting the production of ROS during pathogen attack, which is a key early event in PTI [41]. Its dual role in initiating defense responses while restraining excessive immune activation indicates a finely balanced regulatory mechanism conserved across plant species.
Defense pathways, such as cuticular wax biosynthesis, are conserved across distantly related plant species, shaped by evolutionary pressures from pathogens like Xylella fastidiosa and other xylem-invading microbes. The cuticle acts as a vital barrier, limiting pathogen entry and influencing susceptibility through its composition. Over time, selective pressure from such pathogens has driven the evolution of traits that strengthen plant defenses, including the development of receptor-like kinases (RLKs) that help detect pathogens and regulate protective responses like wax biosynthesis. The widespread conservation of these mechanisms highlights their critical role in enhancing plant survival and reproductive success under pathogen stress [48, 123].
Xylella fastidiosa counters plant resistance network through strategies such as biofilm formation and the production of effectors like oligosaccharides (OPGs). The biofilm, composed of extracellular polymeric substances (EPS), enables Xf to colonize xylem vessels while evading immune detection. This structure also protects the bacteria from environmental stress and interferes with the plant’s ability to mount an effective immune response [81]. In addition to forming biofilms, Xf secretes a range of effectors that disrupt host plant immune responses. OPGs, for example, interfere with both microbe-associated molecular pattern (MAMP)-triggered immunity (MTI) and ETI, weakening the plant’s ability to detect and respond to infection. By suppressing these defense pathways, Xf facilitates its colonization and systemic spread within the host [91]. The density of the bacterium populations critically influences its pathogenic success: at low cell densities, the bacterium adopts a colonization mode to infiltrate the plant, while at high densities it switches to a transmissible form that favours insect vector acquisition—this strategic shift maximizes infection spread while avoiding severe disease symptoms that might deter insect vectors. This dynamic regulation is orchestrated by quorum sensing via diffusible signal factor (DSF), which controls adhesin expression and biofilm formation—key to the switch between plant colonization and insect uptake. Notably, the interplay of biofilm maturation and effector molecules like OPGs represents a sophisticated strategy enabling Xf to suppress host defenses, maintain stealthy persistence, and ensure efficient propagation across both plant and insect hosts [44].
Limitations and future scope
Although the cross-species analysis reveals conserved transcriptional patterns that may underlie common responses to Xylella fastidiosa infection, several factors call for discussion due to methodological constraints inherent to this approach. Variability in RNA-Seq data quality—ranging from sequencing depth to library preparation protocols—can further complicate interpretation by introducing dataset-specific artifacts. The choice of reference genome exerts a notable influence on alignment efficiency and transcript quantification, which in turn can alter the composition of DEGs. Outcomes are also sensitive to the statistical thresholds employed; altering the cutoffs for adjusted p-value or log2 fold change may shift the balance between sensitivity and specificity in DEG detection. Notably, the studies included here utilized different strains of Xf for infection, which likely contributed to heterogeneity in host response. A more unified experimental design using a single, well-characterized strain could enhance the resolution and reliability of conserved transcriptional signals.
To build upon the current findings, future research should prioritize the functional validation of identified hub genes, particularly BAK1,* PED1*,* BAS*,* PIC30*, and WRKY33 which have emerged as central components in the plant immune response network. Techniques such as quantitative real-time PCR and RNA interference can be employed to evaluate their roles in resistance to Xf. To directly assess the contribution of these genes to resistance against Xf, CRISPR/Cas9-based gene editing represents a powerful and precise approach for targeted functional genomics. The application of CRISPR/Cas9 in plant systems has been successfully demonstrated in numerous crops for validating defense-related genes, highlighting its potential utility in this context [27, 54]. By generating knockout or knock-in mutations in BAK1,* PED1*,* BAS*,* PIC30*, and WRKY33 researchers can investigate the phenotypic consequences of gene disruption in planta, providing definitive evidence for their roles in Xf resistance or susceptibility.
Final remarks
The analysis identified 18 differentially expressed genes (DEGs) linked to resistance against Xylella fastidiosa, including BAK1,* PED1*,* BAS*,* PIC30*, WRKY33, VEIN PATTERNING 1 and WRKY40, pivotal for immune responses and plant resilience. Their consistent expression across host plants suggests a shared resistance network. Notably, the protein-protein interactions among these DEGs form a complex regulatory framework crucial for understanding the plant’s defense mechanisms. This network represents a sort of in silico scenario, where computational models can predict and validate the functional roles of these genes in immunity. These findings are instrumental for future research on plant resistance, guiding the functional validation of these genes and informing breeding programs for resistant cultivars, especially in olives and grapes. Future studies should investigate the roles of these genes in immunity and their interactions within signaling networks. Genomic technologies like CRISPR/Cas9 hold promise in the future to enhance resistance traits, and combining these with traditional breeding is essential for addressing Xylella fastidiosa challenges. Ultimately, integrating genomic data with breeding strategies will help develop resilient crops, enhancing food security and promoting sustainable agriculture.
Supplementary Information
Supplementary Material 1.
Supplementary Material 2.
Supplementary Material 3.
Supplementary Material 4.
Supplementary Material 5.
Supplementary Material 6.
Supplementary Material 7.
Supplementary Material 8.
Supplementary Material 9.
Supplementary Material 10.
Supplementary Material 11.
Supplementary Material 12.
Supplementary Material 13.
Supplementary Material 14.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bernal AJ, Jensen JK, Harholt J, Sørensen S, Moller I, Blaukopf C, Johansen B, de Lotto R, Pauly M, Scheller HV, Willats WG. Disruption of ATCSLD 5 results in reduced growth, reduced xylan and homogalacturonan synthase activity and altered xylan occurrence in Arabidopsis. Plant J. 2007;52(5):791–802. 10.1111/j.1365-313X.2007.03281.x. PMID: 17892446.10.1111/j.1365-313X.2007.03281.x 17892446 · doi ↗ · pubmed ↗
- 2Broekaert WF, Delauré SL, De Bolle MF, Cammue BP. The role of ethylene in host-pathogen interactions. Annu Rev Phytopathol. 2006;44:393–416. 10.1146/annurev.phyto.44.070505.143440. PMID: 16602950.10.1146/annurev.phyto.44.070505.14344016602950 · doi ↗ · pubmed ↗
- 3Castro C, Di Salvo B, Roper MC. Xylella fastidiosa: a reemerging plant pathogen that threatens crops globally. P Lo S Pathog. 2021;17(9). 10.1371/journal.ppat.1009813.10.1371/journal.ppat.1009813 PMC 842856634499674 · doi ↗ · pubmed ↗
- 4Chen H, De La Fuente L. Calcium transcriptionally regulates movement, recombination and other functions of Xylella fastidiosa under constant flow inside microfluidic chambers. Microb Biotechnol. 2020 Mar;13(2):548-561. Epub 2019 Nov 14. PMID: 31729188; PMCID: PMC 7017821. 10.1111/1751-7915.1351210.1111/1751-7915.13512 PMC 701782131729188 · doi ↗ · pubmed ↗
- 5Chen K, Gao J, Sun S, Zhang Z, Yu B, Li J, Xie C, Li G, Wang P, Song CP, Bressan RA, Hua J, Zhu JK, Zhao Y. BONZAI proteins control global osmotic stress responses in plants. Curr Biol. 2020;30(24):4815–25.e 4. PMID: 33035480. 10.1016/j.cub.2020.09.01610.1016/j.cub.2020.09.01633035480 · doi ↗ · pubmed ↗
- 6Dielen AS, Badaoui S, Candresse T, German-Retana S. The ubiquitin/26S proteasome system in plant-pathogen interactions: a never-ending hide-and-seek game. Mol Plant Pathol. 2010;11(2):293–308. 2009.00596.x. Erratum in: Mol Plant Pathol. 2011;12(1):103. PMID: 20447278; PMCID: PMC 6640532.10.1111/j.1364-3703.2009.00596.x PMC 664053220447278 · doi ↗ · pubmed ↗
- 7Eh TJ, Jiang Y, Jiang M, Li J, Lei P, Ji X, Kim HI, Zhao X, Meng F. The role of trehalose metabolism in plant stress tolerance. J Adv Res. 2024;S 2090-1232(24)00603-9. 10.1016/j.jare.2024.12.025. PMID: 39708962.10.1016/j.jare.2024.12.02539708962 · doi ↗ · pubmed ↗
- 8Federici MT, Marcondes JA, Picchi SC, Stuchi ES, Fadel AL, Laia ML, Lemos MVF, Macedo Lemos EG. Xylella fastidiosa: An in vivo system to study possible survival strategies within citrus xylem vessels based on global gene expression analysis. Electron J Biotechnol [Internet]. 2012 May 11 [cited 2025 Jul 31];15(3). https://www.ejbiotechnology.info/index.php/ejbiotechnology/article/view/v 15n 3-4