A Case of Myxoid Malignant Peripheral Nerve Sheath Tumor in a Patient With Carney Complex
Kazuhiro Kobayashi, Natsuko Suzui, Hirofumi Shibata, Takenori Ogawa, Tatsuhiko Miyazaki

TL;DR
This paper reports the first case of a patient with Carney Complex developing a myxoid malignant peripheral nerve sheath tumor in the salivary gland.
Contribution
The novelty lies in identifying a myxoid malignant peripheral nerve sheath tumor in a Carney Complex patient, a previously unreported association.
Findings
A patient with Carney Complex was diagnosed with a myxoid low-grade malignant peripheral nerve sheath tumor in the parotid gland.
The tumor exhibited S100+, CD34+, SOX10+, and MIB-1 positivity, with a myxomatous stroma and cribriform pattern.
This case represents the first known instance of such a tumor arising in a CNC patient.
Abstract
Background: Carney complex (CNC) is a group of disorders characterized by endocrine hyperactivity or tumors, abnormal skin pigmentation, myxomas of the skin and heart, and adrenocortical and pituitary tumors; in most cases, the disorder is inherited in an autosomal dominant manner. Case Presentation: We, herein, report a female patient who had undergone a total of seven left parotid tumor resections since the age of 45 years. At age 50, genetic testing confirmed a c.597del C (p. Phe200LeufsX6) mutation in the type-1α regulatory subunit of cAMP-dependent protein kinase (PRKAR1A); this led to a diagnosis of CNC for the patient and the patient's second and third daughters. At the age of 55, the left parotid gland became rapidly enlarged, and surgery was performed because recurrence was suspected. Intraoperative rapid pathological diagnosis revealed a mucous tumor with an unknown…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
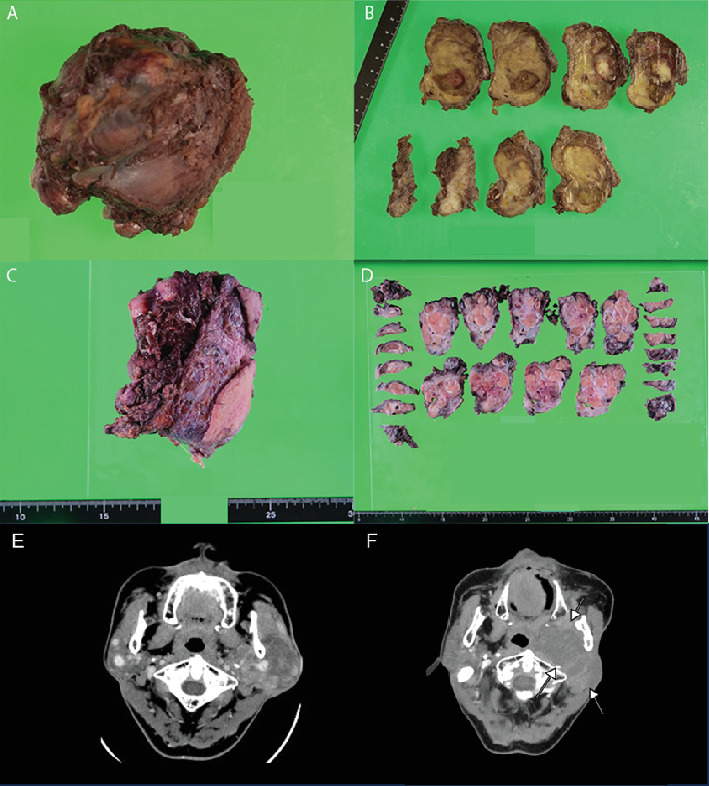 Figure 1
Figure 1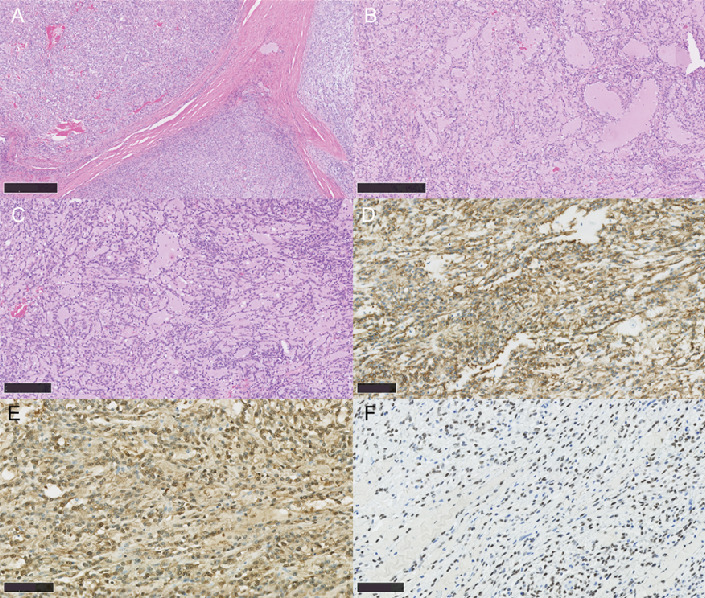 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiac tumors and thrombi · Neurofibromatosis and Schwannoma Cases · Sarcoma Diagnosis and Treatment
1. Introduction
Carney complex (CNC) is a group of disorders characterized by endocrine hyperactivity or tumors, abnormal skin pigmentation, myxomas of the skin and heart, and adrenocortical and pituitary tumors; in most cases, the disorder is inherited in an autosomal dominant manner. Mutations in the type-1α regulatory subunit of cAMP-dependent protein kinase (PRKAR1A) are implicated in the development of CNC.
MPNSTs typically affect adults with a mean age of 50.7 and a male-to-female ratio of 1.5:1. The prevalence for males has been reported in both sporadic and NF1-associated MPNST contexts. Head and neck MPNSTs have been reported in various sites, such as the cheek, tongue, infratemporal fossa, brachial plexus, paranasal sinus, orbit, and parotid gland [1]. However, primary intraosseous tumors are extremely rare, with only a few cases documented in the English-language literature [2–7].
Here, we report a case of CNC diagnosis in a mother and two of her daughters; the mother developed a myxoid MPNST derived from the salivary glands, which to our knowledge has not previously been reported.
2. Case Presentation
2.1. Medical History
Left parotid tumor resection, pituitary tumor removal (GHoma [growth hormone–producing pituitary adenoma]), laparoscopic salpingo-oophorectomy (serous cystadenoma), and multiple cardiac tumor resections (myxoma).
2.2. Current Medical History
The female patient has undergone a total of seven times of left parotid tumor resections since the age of 45 years. At age 45, she underwent surgery for a left parotid tumor at another hospital. At that time, she was diagnosed with a benign tumor (details unknown). At the age of 50, genetic testing of the recurrent tumor confirmed a c.597del C (p. Phe200LeufsX6) mutation in the PRKAR1A; the patient and the patient's second and third daughters were diagnosed with CNC [8, 9]. At the age of 55, the residual left parotid gland rapidly enlarged, and surgery was performed. Intraoperative rapid pathological diagnosis revealed a mucinous tumor with an unknown differentiation grade; tumor was resected at our hospital. The resected tumor at that time manifested S100^+^, CD34^+^ mucinous spindle cell tumor (Figures 1A, 1B, and 1E). Although we considered the lesion of myoepithelioma or solitary fibrous tumor (SFT), neither was considered typical in this case. Due to metastasis to the lymph node and involvement of the submandibular gland, the patient was followed up as having a low-grade malignant atypical myxoid tumor. At age 60, surgical intervention for a tumor recurrence revealed invasion of the temporal muscle, with a malignancy of the same histologic type as the previously resected. After that, recurrences were seen in the lymph node and parapharyngeal stromal at age 63, in the parotid gland at age 65, and a parapharyngeal space at age 68 with the same pathological manifestations. The patient died at 73 years of age due to systemic metastasis of the tumor.
2.3. Family History
Family history is as follows: The second daughter was diagnosed with CNC, had facial and lip pigmentation, and underwent laparoscopic bilateral adrenalectomy. The third daughter was also diagnosed with CNC, had facial and lip pigmentation, and underwent laparoscopic bilateral adrenalectomy. She was also found to have multiple myxomas in the left atrium.
2.4. Gross Pathology
A 7 cm × 5 cm yellow lobulated mass was seen. The tumor invaded the surrounding muscle tissue (Figures 1C, 1D, and 1F).
2.5. Histological Findings
Tumor cells were uniformly and somewhat sparsely distributed, and the lesions were generally nodular with a fibrous capsule surrounding them, but some had invasion into the surrounding area. The interior showed fibrous septa. The growth of spindle-shaped or linear atypical cells in a sheet-like, cord-like, partially saccular, or cribriform pattern was observed, with abundant myxomatous matrix deposition in the stroma. The atypical cells had a high nuclear/cytoplasm ratio: nuclei with nucleoli, a well-defined nuclear border, and increased fine chromatin. The cytoplasm was predominantly acidophilic but was accompanied by granular basophilic structures in some places. Infiltration of neutrophils, lymphocytes, and plasma cells was observed. The stroma was partially accompanied by atrophied salivary gland tissue. The tumor had also invaded the surrounding musculature and adipose tissues. Immunohistochemistry showed that tumor cells were S100^+^, CD34^+^, SOX10^+^, c-kit^−^, desmin^−^, DOG1^−^, ER^−^, HMB45^−^, STAT6^−^, Oct4^−^, synaptophysin^−^, AE1/AE3^−^, SMA^−^, and vimentin^+^; for a small fraction of cells, EMA^+^, synaptophysin^−^, BCL2^−^, and β-catenin^−^ and MIB-1 positivity was about 1% (Figure 2 and Table 1). Based on these results, this case was diagnosed as myxoid low-grade MPNST.
3. Discussion
CNC is a disease characterized by the development of skin pigmentation, endocrine hyperactivity, adrenocortical tumors, pituitary adenomas, and cardiac myxomas [10, 11]. Currently, CNC is diagnosed according to the diagnostic criteria listed in Table 2. According to Stratakis et al., the diagnostic criteria for CNC require either two or more major manifestations—such as spotty skin pigmentation, cardiac myxoma, or endocrine tumors—or one major manifestation along with a supplemental criterion, such as a PRKAR1A mutation or an affected first-degree relative. These criteria are widely accepted and have been used in the evaluation of this patient [12]. In the case presented here, about half of the cardiac myxomas and CNCs classified in the major criteria are inherited in an autosomal manifest manner. While 70% of patients diagnosed with CNC are known to be hereditary, about 30% are considered to be nonhereditary mutations. CNC is known to be complicated by a variety of tumors and symptoms.
Furthermore, rare manifestations of CNC continue to be reported, expanding the clinical spectrum of the disease. For example, Savva et al. described a case of bilateral myxoid fibroadenomas of the breast in a CNC patient, emphasizing the variability in tumor presentation and the need for more comprehensive surveillance in affected individuals [13]. Such cases underscore the importance of accumulating clinical data to better define the natural history of CNC and improve diagnostic strategies.
Our survey of the CNC literature up to 2022 revealed only limited reports of malignancies; these included thyroid cancer, acinar cell carcinoma and adenocarcinoma of the pancreas, and colonic and gastric carcinoma (Table 3). Reports of myxoid malignant soft-tissue tumors are minimal. Psammomatous melanotic schwannoma (PMS) is a rare tumor of the central and peripheral nervous system that occurs in some CNC patients [14, 15]; the tumor is multicentric with frequent calcification and pigmentation [16, 17]. The most frequent sites of PMS are the gastrointestinal tract and paraspinal sympathetic chain [18]. Spinal cord tumors occur in adults (average age 32 years) and generally manifest as painful lesions with nerve root involvement. Approximately 10% of CNC-associated PMS go on to become malignant [19]. This tumor was renamed “malignant melanotic nerve sheath tumor” in the 5th edition of the WHO Classification of Tumors of Soft Tissue and Bone [20]. In our present case, there was no melanin deposition or sandy calcification deposits, which are considered characteristic of PMS. Therefore, we considered the tumor to be a malignancy of the nervous system that had not been previously reported to occur in association with CNC. In addition, we believe that this tumor should be categorized as a myxoid MPNST because it recurred locally and metastasized to the lung.
More than 70% of patients diagnosed with CNC carry mutations in the PRKAR1A gene (the “CNC1 locus”); the frequency increases to 80% in patients with Cushing syndrome due to primary pigmented nodular adrenocortical disease (PPNAD) [21, 22].
To date, 401 unrelated CNC families of diverse ethnic origins have been reported [21, 23, 24]. Pathogenic mutations in PRKAR1A include single nucleotide substitutions, small (≤ 15 bp) deletions/insertions, complex reorganizations that span the entire open reading frame of the gene, and large deletions that cover most exons [15]. Most of these mutations are unique to a single family, with only three pathological variants (c.82C>T, c.491_492delTG, and c.709-2_709-7 delATTTTT) identified in more than three unrelated families [15, 21, 25].
The PRKAR1A mutations associated with CNC lead to loss of function of the R subunit, which normally inhibits the catalytic C subunit of PKA. The activity of the “uninhibited” C subunit leads to increased phosphorylation of the downstream PKA substrate, cAMP response–binding protein (CREB), increased cell proliferation in cAMP-responsive tissues, and tumor formation in CNC-affected tissues [26, 27].
It is challenging to predict the molecular pathological mechanisms that directly link CNC and MPNST due to the absence of case reports connecting them directly. However, it is hypothesized that Protein Kinase A (PKA) typically acts to suppress the MAPK pathway in normal cells. In cells with PRKAR1A mutations, where regulatory subunit Type 1A is either absent or the PRKAR1A mutation is ineffective, an overproduction of other regulatory subunits (mainly Type II PKA) or a defect in Type I PKA may occur. These changes in CNC cells could relatively activate the extracellular signal-regulated kinase in the MAPK pathway, potentially leading to abnormal proliferation and growth.
The development of MPNST has been reported to involve the activation of a part of the classical MAPK pathway, the RAS-RAF-MEK pathway [28]. These mechanisms may have caused the development of MPNST in this patient. However, many aspects, such as malignancy, cannot be explained by these mechanisms alone, and further examination is awaited.
In a recurrent tumor in the parapharyngeal space of the patient discussed in our report, ARID1A G2087R, PRKR1A F200fs∗6, and RAD21 amplification were identified as disease-relevant alterations by genetic testing. PRKAR1A gene mutations are included among the supplemental criteria used to make a diagnosis of CNC (Table 1). How the PRKAR1A mutation detected in this CNC is related to tumorigenesis is currently unclear.
Prkar1a +/− mice develop nonpigmented schwannomas and fibrotic bone lesions beginning at about 6 months of age; 10% of the mice subsequently develop thyroid tumors [29]. Another mouse model with significantly more Prkar1a downregulation and significantly higher cAMP signaling produced a more severe CNC phenotype [30]. Although reported in Prkar1a heterozygous mice, the presence of nonpigmented schwannomas is consistent with our conclusion that the cancer is a mucinous malignant neurogenic tumor arising in association with CNC. Careful clinical observation and documentation of future CNC patients will help to substantiate our claims.
4. Conclusion
We have identified a previously unreported myxoid malignant nervous system tumor that developed in a patient with CNC.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abdel Razek A. A. K. Gamaleldin O. A. Elsebaie N. A. Peripheral Nerve Sheath Tumors of Head and Neck: Imaging-Based Review of World Health Organization Classification Journal of Computer Assisted Tomography 202044692894010.1097/RCT.000000000000110933196600 · doi ↗ · pubmed ↗
- 2Che Z. Nam W. Park W. S. Intraosseous Nerve Sheath Tumors in the Jaws Yonsei Medical Journal 20064722642701664255910.3349/ymj.2006.47.2.264PMC 2687639 · doi ↗ · pubmed ↗
- 3Cunha J. L. S. Tomo S. de Paiva Gomes Fernandes E. K. Primary Intraosseous Malignant Peripheral Nerve Sheath Tumor of the Mandible: An Unusual Presentation Mimicking a Benign Lesion Oral Oncology 202112010526610.1016/j.oraloncology.2021.10526633810988 · doi ↗ · pubmed ↗
- 4Kim H. Y. Hwang J. Y. Kim H. J. CT, MRI, and (18)F-FDG PET/CT Findings of Malignant Peripheral Nerve Sheath Tumor of the Head and Neck Acta Radiologica 201758101222123010.1177/02841851166846742-s 2.0-8502746080628068826 · doi ↗ · pubmed ↗
- 5Lee S. Lee C. Kim J. K. Nam W. An Unusual Presentation of Intraosseous Malignant Peripheral Nerve Sheath Tumour of Mandible Dentomaxillofacial Radiology 20194872018034110.1259/dmfr.201803412-s 2.0-8507250888731188646 PMC 6775785 · doi ↗ · pubmed ↗
- 6Sham M. E. Ghorpade A. Shetty S. Hari S. Vinay Malignant Peripheral Nerve Cell Tumour Journal of Maxillofacial and Oral Surgery 201091687110.1007/s 12663-010-0019-62-s 2.0-8490344635723139572 PMC 3453695 · doi ↗ · pubmed ↗
- 7Zakhary I. Elsalanty M. Ishag I. Malignant Peripheral Nerve Sheath Tumor of Mandible Journal of Craniofacial Surgery 201122276276610.1097/SCS.0b 013e 318207 f 4472-s 2.0-7995387538621415661 · doi ↗ · pubmed ↗
- 8Ishida N. Shimabukuro K. Matsuno Y. Arakawa Y. Takemura H. Tumorectomy With Right Thoracotomy for Synchronous Left Atrial Myxomas From Carney Complex: Report of a Case Surgery Today 201444118518710.1007/s 00595-012-0369-42-s 2.0-8489174373423052755 · doi ↗ · pubmed ↗