Case Report: De novo DHX37 mutations in Saudi patients with 46,XY differences of sex development
Abeer Alabduljabbar, Sara Abid, Dania Farooq, Sara Aljazaeri, Yara Khamag, Raghad Alhuthil, Latifah Alfahad, Afaf Alsagheir

TL;DR
This paper reports on three Saudi patients with 46,XY differences of sex development caused by mutations in the DHX37 gene, highlighting the gene's role in testicular development.
Contribution
The study presents the first case series of DHX37-related DSD in Saudi Arabia and identifies a novel mutation.
Findings
Three Saudi patients with 46,XY DSD were found to have DHX37 mutations, including one novel variant.
Phenotypic variability was observed, ranging from complete testicular regression to partial gonadal dysgenesis.
Genetic testing, especially whole-exome sequencing, is emphasized for accurate diagnosis in high-consanguinity regions.
Abstract
Differences of sex development (DSD) are a group of congenital conditions involving atypical chromosomal, gonadal, or anatomical sex development. DHX37, a gene involved in ribosome biogenesis, located on chromosome 12, at the 12q24.31 region, has recently emerged as a contributor to 46,XY DSD, particularly gonadal dysgenesis and testicular regression syndrome (TRS). This study presents a case series from Saudi Arabia highlighting novel and known DHX37 variants in three patients with 46,XY DSD. Three Saudi patients presented with ambiguous genitalia, non-palpable or atrophic testes, and hypergonadotropic hypogonadism. Identified variants included two known (p.Arg308Gln, p.Arg674Trp) and one novel (p.Gly478Val) missense mutation. Phenotypic variability ranged from complete testicular regression to partial gonadal dysgenesis. Thus, this is the first case series of DHX37-related DSD in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
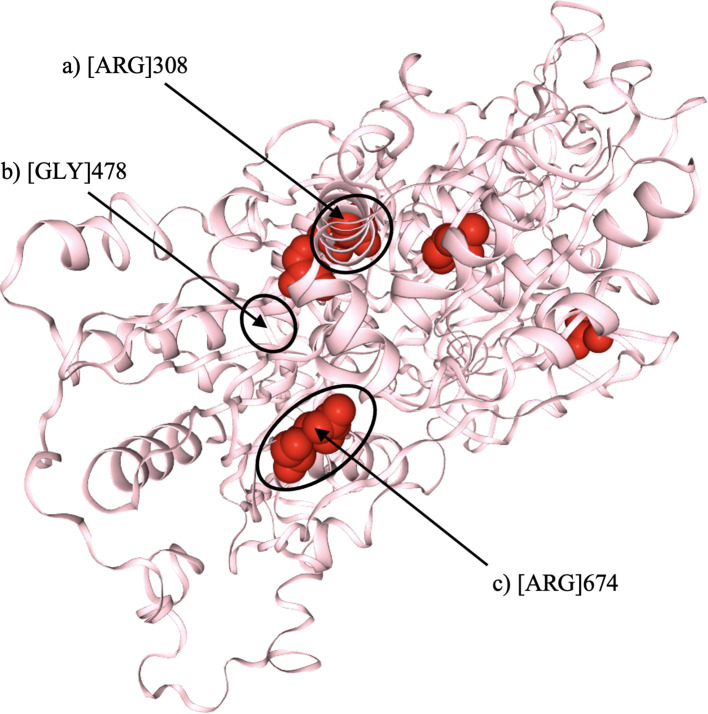 Figure 1
Figure 1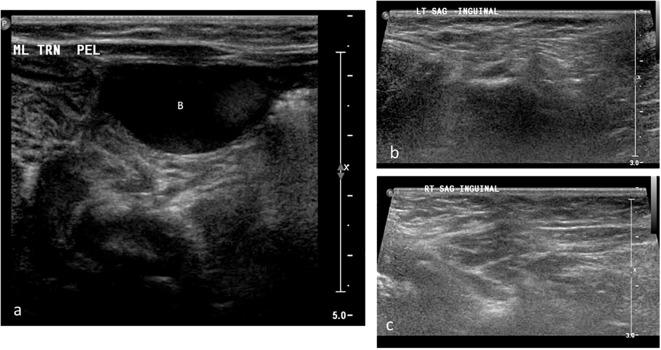 Figure 2
Figure 2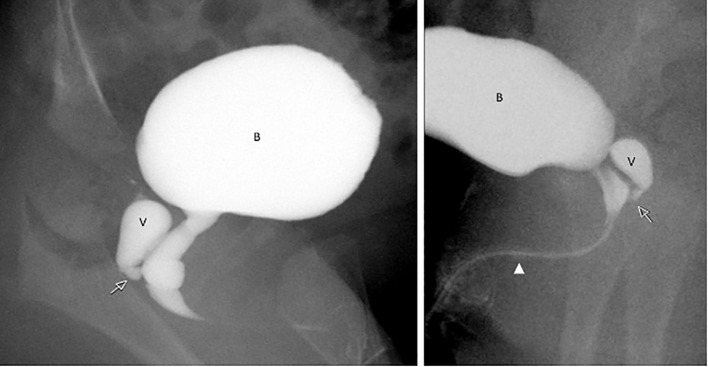 Figure 3
Figure 3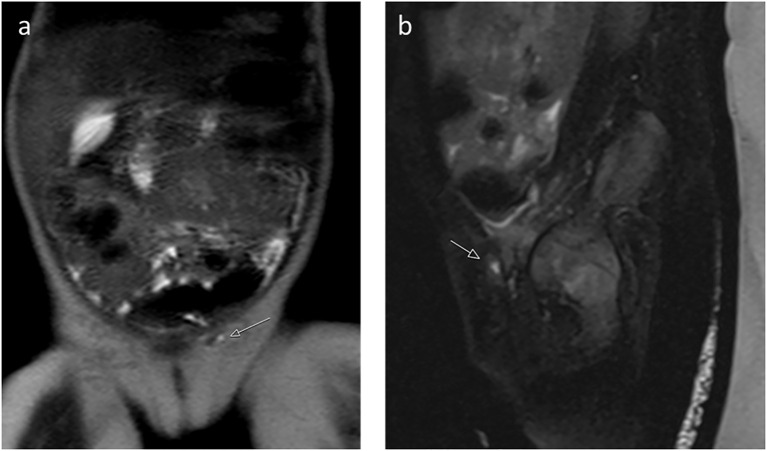 Figure 4
Figure 4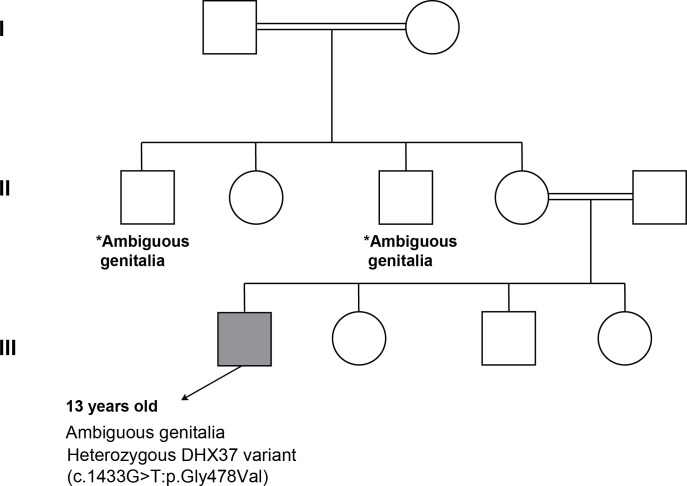 Figure 5
Figure 5| Feature | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| Age of presentation and the age of hormonal testing | 2 years | 9 years | 2 years |
| Assigned gender at birth | Female | Male | Male |
| Karyotype | 46,XY | 46,XY | 46,XY |
| Gonadal palpability | Non-palpable | Non-palpable | Non-palpable |
| Phenotype | TRS | TRS | PGD |
| Genital findings | Phallus-like, posterior labial fusion, single opening | Micropenis, fused labioscrotal folds, single opening | Phallus 0.5 cm, fused labioscrotal folds, single opening |
| Imaging findings | Absence of both testicular tissue or only atrophic gonadal remnants | Absence of functional testicular tissue with only remnants present | Presence of a structure resembling testicular tissue in the inguinal region |
| Family history | Negative | Negative | Positive (maternal uncles) |
| FSH (IU/L) | 96.8 | 59.2 | 54.2 |
| LH (IU/L) | 5.0 | 25.2 | 23 |
| Androstenedione (nmol/l) | < 1 | < 1 | < 1 |
| Testosterone (nmol/l) | < 0.10 | < 0.10 | < 0.10 |
| DHT (pg/ml) | < 50 | < 50 | < 50 |
| DHEA (ng/ml) | < 0.5 | < 0.5 | < 0.5 |
| Sex assignment | Reassigned to male | Continued male | Continued male |
| Surgical intervention | Surgical correction, orchidopexy | Bilateral orchiectomy | Orchidopexy |
| Hormonal therapy | GH + Testosterone | Testosterone | Testosterone |
| Age at last follow-up | 15 years | 13 years | 13 years |
| Case | DNA change | Protein change | Exon | dbSNP ID | Population frequency | Conservation |
| ACMG classification | Inheritance |
|---|---|---|---|---|---|---|---|---|---|
| 1 | c.923G > A | p.Arg308Gln | 6 | rs1384892917 | Extremely rare (< 0.0001) | Highly conserved across species | SIFT: Deleterious | Pathogenic ( | Heterozygous, |
| 2 | c.2020C > T | p.Arg674Trp | 15 | rs1954336272 | Absent in gnomAD | Highly conserved (arginine critical in helicase domain) | SIFT: Deleterious | Likely pathogenic ( | Heterozygous, |
| 3 | c.1433G > T | p.Gly478Val | 11 | Not reported (novel) | Not in gnomAD/dbSNP | Highly conserved glycine residue | SIFT: Deleterious | VUS | Heterozygous, |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSexual Differentiation and Disorders · Genetic and Clinical Aspects of Sex Determination and Chromosomal Abnormalities · Urological Disorders and Treatments
Introduction
1
Differences of sex development (DSD) encompass a group of congenital conditions characterized by atypical development of chromosomal, gonadal, or anatomical sex (1). Among these, individuals with a 46 XY karyotype may present with varying degrees of undervirilization, including ambiguous genitalia or incomplete masculinization (2). This spectrum includes complete and partial gonadal dysgenesis (CGD, PGD) and testicular regression syndrome (TRS) (3, 4).
TRS is a rare subtype of 46,XY DSD marked by partial or complete absence of testicular tissue in one or both gonads, despite the presence of the 46,XY (5). It affects approximately 1 in 20,000 boys and is often attributed to prenatal testicular loss, possibly due to vascular accidents (6). Diagnosis typically requires a multidisciplinary approach involving clinical evaluation, hormonal profiling, imaging, cytogenetic testing, and, in some cases, surgical exploration (6).
The genetic basis of 46,XY DSD is becoming increasingly well understood, with several genes now recognized as key regulators of testicular differentiation and gonadal development. Among these, DEAH-box RNA helicase (DHX37) has emerged as a notable contributor, particularly in cases of 46,XY gonadal dysgenesis and TRS. Initially, DHX37 mutations, located on chromosome 12, at the 12q24.31 region (7), were reported in syndromic neurodevelopmental disorders involving microcephaly, global developmental delay, cardiac and renal anomalies, and seizures (8–10). However, more recent findings have demonstrated its role in non-syndromic DSD, with pathogenic heterozygous missense variants linked to a wide phenotypic spectrum—from phenotypic females to males with undescended or atrophic testes (10, 11).
Currently, 21 distinct DHX37 variants have been reported in 58 individuals with 46,XY DSD, spanning a wide phenotypic spectrum—from phenotypic females to males with undescended or atrophic testes (12). The gene plays a central role in ribosome biogenesis, and its mutations are thought to specifically impair gonadal development without broader systemic involvement (8, 9).
Here, we present a case series of three patients with 46,XY DSD from a tertiary care center in Saudi Arabia, each carrying heterozygous DHX37 missense variants, including one novel de-novo mutation (p.Gly478Val). This report expands the genotypic and phenotypic spectrum of DHX37-related DSD and underscores the value of genetic testing in elucidating the etiology of 46,XY DSD—particularly in populations with high consanguinity, such as Saudi Arabia, where the prevalence of rare genetic disorders is elevated (13).
Methodology
2
Over the past 10 years (2014–2024), our institution’s endocrinology clinic evaluated 500 patients with various types of DSD. Among them, genetic testing identified three individuals with pathogenic variants in the DHX37 gene associated with 46,XY DSD.
Genetic testing was conducted as part of routine diagnostic evaluation for all three patients. Genomic DNA was extracted from peripheral blood samples, and whole-exome sequencing (WES) was performed using the Illumina HiSeq2500 platform with an average target depth of 80×. DNA libraries were prepared using the Agilent SureSelect All Exons V6 (50 Mb) capture kit.
Identified variants were assessed and classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines (14), using the Varsome database (15).
To further investigate the structural impact of the identified mutations, in-silico protein modeling was conducted using SwissModel, based on Protein Data Bank (PDB) entry 7d4i, chain RZ (Figure 1) (16).
Three-dimensional structural model of the DHX37 protein, illustrating the locations of the three identified variants (indicated by arrows). (a) Substitution of arginine (Arg) at position 308 with glutamine (p.Arg308Gln). (b) Substitution of glycine (Gly) at position 478 with valine (p.Gly478Val). (c) Substitution of arginine (Arg) at position 674 with tryptophan (p.Arg674Trp). All three variants are predicted to disrupt hydrogen bonding within conserved helicase domains, potentially impairing protein function. Structural modelling was performed using SwissModel (PDB ID: 7d4i, chain RZ).
Ethical approval was obtained from the ethics committee at KFSHRC (reference: 2231110), and written consent was taken from all patients’ legal guardians.
Results
3
A summary of clinical, biochemical, and genetic features is presented in Tables 1, 2.
Patient 1
3.1
A 2-year-old, initially raised as female, was referred for evaluation of ambiguous genitalia. Examination revealed a phallus-like structure, posterior labial fusion, and a single perineal opening. The karyotype was 46,XY with a positive SRY gene. Hormonal evaluation showed hypergonadotropic hypogonadism and poor androgen response to hCG stimulation, suggesting primary gonadal failure.
Imaging revealed no uterus or ovaries. Ultrasound showed absence of testicular tissue (Figure 2). A genitogram demonstrated an elongated urethra with posterior outpouching (Figure 3), and an MRI showed a male-type urethra with a small vaginal-like structure connecting to the urogenital sinus with a small, high-signal intensity structure in the left inguinal canal, suggestive of undescended gonadal tissue (Figure 4). Cystoscopy showed a prostatic utricle and a single introitus. Laparoscopy identified a malformed right intra-abdominal testis and a left testis near the internal inguinal ring.
Grayscale ultrasound images of the pelvis and bilateral inguinal canals in Patient 1. (a) Transverse pelvic scan showing the urinary bladder (B); no uterus or gonadal structures are visualized posterior to the bladder. (b) Longitudinal scan of the left inguinal canal demonstrating absence of gonadal tissue (arrow). (c) Longitudinal scan of the right inguinal canal also showing no visible gonadal tissue.
Catheter genitogram images from Patient 1 illustrating internal urogenital anatomy. The contrast study shows a vaginal remnant (V) posterior to the urinary bladder (B), joining the elongated urethra at the level of the urogenital sinus (arrow). The urethra is elongated and male like in configuration (arrowhead), consistent with undervirilized external genitalia.
(a) Coronal T2-weighted image and (b) sagittal T2-weighted image with fat saturation reveal a small, high-signal intensity structure in the left inguinal canal, suggestive of undescended gonadal tissue, and showed a male-type urethra with a small vaginal-like structure connecting to the urogenital sinus (Patient 1).
At 3 years of age, the patient was assessed for short stature, with a recorded height of 90.1 cm (–2.64 SD). Growth hormone stimulation testing using clonidine and glucagon yielded a peak GH level of 8.6 ng/ml, leading to the initiation of growth hormone therapy, which resulted in a 10 cm/year growth velocity during the first year. In the second year, growth velocity was 8 cm/year, followed by a sustained rate of 5–6 cm/year in subsequent years. At age 13 years, whole exome sequencing identified a de-novo heterozygous pathogenic DHX37 variant (c.923G > A:p.Arg308Gln). This patient was diagnosed with TRS. Following a multidisciplinary team evaluation—including pediatric endocrinology, psychology, urology, and ethics consultation—the family received comprehensive counseling on the underlying diagnosis, prognosis, and available management options, including potential gender identity development, fertility implications, and surgical outcomes. After several sessions and with psychological support, the family elected to proceed with male gender assignment. The patient was initiated on testosterone replacement therapy to promote virilization. Subsequently, he underwent staged corrective genital surgery, including hypospadias repair and scrotoplasty. Postoperatively, he showed good surgical outcomes, with appropriate penile appearance and urinary function. At the last follow-up, both the patient and family expressed satisfaction with the decision and treatment outcomes, and the patient has shown stable psychosocial adaptation in the male gender role.
Patient 2
3.2
A 9-year-old child, raised as male, presented with ambiguous genitalia; otherwise healthy, his karyotype analysis revealed a 46,XY chromosomal pattern. Physical examination showed a small phallus, fused labioscrotal folds, and non-palpable gonads. Hormonal evaluation demonstrated low baseline testosterone and gonadotropin levels, with no significant response to human chorionic gonadotropin (hCG) stimulation testing. Pelvic imaging did not reveal müllerian structures or definitive testicular tissue but identified a prostatic utricle and structures suggestive of atrophic testes.
Laparoscopy confirmed the presence of bilateral intra-abdominal atrophic testicular remnants, consistent with TRS. Genetic testing identified a de-novo heterozygous likely pathogenic DHX37 variant (c.2020C > T:p.Arg674Trp), further supporting the diagnosis. He received four doses of testosterone at the age of 9 years old. After the testosterone injection, the patient started to have a hoarse voice, he got taller, and he started to have body, pubic, and armpit hair. Given the non-functional nature of the intra-abdominal gonadal remnants and the increased risk of malignant transformation, a decision was made to proceed with bilateral orchiectomy at 11 years of age. The patient continued to be assigned and raised as male, with ongoing psychological support and endocrine follow-up to address pubertal development and long-term care. At the most recent follow-up, he appeared clinically well and vitally stable. Examination revealed pubic hair, a small stretched phallus measuring 1 cm, a single perineal opening, fused labioscrotal folds resembling a scrotum, and an absence of palpable testes. No additional health concerns were reported.
Patient 3
3.3
A 2-year-old male presented with ambiguous genitalia, primary gonadal failure, and a positive family history of similar cases in two maternal uncles (Figure 5), although further details regarding their conditions were unavailable. Physical examination revealed a micropenis, fused labioscrotal folds, and a single perineal opening. Laboratory evaluation at 2 years of age showed elevated gonadotropin levels, and human chorionic gonadotropin (hCG) stimulation testing demonstrated a poor androgen response, consistent with primary gonadal failure. Imaging studies identified an inguinal structure consistent with testicular tissue.
*Pedigree analysis of family of Patient 3. Both maternal uncles had ambiguous genitalia (no identified etiology, segregation analysis was done for both parents and was negative).
At the initial presentation to our clinic, the patient underwent biochemical evaluation followed by genetic testing, which initially yielded negative results. Initially, the patient was diagnosed with partial gonadal dysgenesis at the age of 2 years. After multidisciplinary discussions with the family, a decision was made to raise the patient as a male and assign a male gender. By the age of 3 years, he received testosterone to enhance phallic growth, and by the age of 4 years, he underwent bilateral orchidopexy. However, based on strong clinical suspicion of an underlying genetic etiology, a repeated genetic test identified a novel variant of uncertain significance (VUS), the DHX37 variant (c.1433G > T; p.Gly478Val). Parental testing confirmed the absence of the variant, supporting its de-novo origin. He is currently under regular follow-up. Testosterone replacement therapy is planned at the appropriate time to induce pubertal development. The patient is reported to be doing well, attending school regularly, and has no additional health concerns. The patient remains under regular follow-up to monitor development and assess the potential risk of gonadal malignancy.
Discussion
4
In this case series, we identified three Saudi patients with heterozygous missense variants in DHX37: p.Arg308Gln, p.Arg674Trp, and a novel variant, p.Gly478Val. All variants affect highly conserved residues within functional domains, likely altering helicase activity and interactions with RNA or associated cofactors essential for ribosome assembly (9). Each patient had a 46,XY karyotype and presented with ambiguous genitalia, non-palpable or atrophic testes, and hypergonadotropic hypogonadism—consistent features of underlying gonadal dysfunction.
This is the first report of DHX37-related DSD in the Saudi population, expanding the gene’s mutational spectrum and emphasizing the importance of population-specific genetic studies (9). With the high rates of consanguinity in the region (13), investigating such rare variants is critical for improving diagnostic accuracy and informing genetic counseling.
None of the patients exhibited intellectual or developmental delay, aligning with previous findings that DHX37-related DSD is typically non-syndromic (9). Patient 1 carried the c.923G > A (p.Arg308Gln) variant, classified as pathogenic (17), and repeatedly associated with TRS as reported previously in five affected patients that had gonadal dysgenesis phenotype (7). Patient 2 had the p.Arg674Trp variant, which is classified as likely pathogenic (18), and affects a conserved residue and likely compromises helicase function (7). Patient 3 was found to carry a novel p.Gly478Val variant. Although it was classified as VUS according to ACMG guidelines (14) in the laboratory report, the patient was clinically diagnosed with PGD.
Phenotypic variability among DHX37 mutation carriers is well documented and was evident in our series. While Patient 1 presented with complete gonadal regression, Patient 3 showed partial dysgenesis with some residual testicular tissue. This variability supports the hypothesis that additional genetic modifiers, epigenetic influences, or environmental factors may contribute to disease expression (8). Furthermore, the family history of genital ambiguity in Patient 3 raises the possibility of germline mosaicism, a phenomenon previously described in other DSD-associated gene (19, 20).
These cases highlight the clinical utility of WES in diagnosing DSD, particularly when hormonal and imaging studies are inconclusive or yield overlapping findings. In Patient 1, genetic confirmation of a DHX37 mutation supported gender reassignment and initiation of testosterone therapy. In Patient 2, the identification of a pathogenic DHX37 variant contributed to the decision for bilateral orchiectomy due to the presence of non-functional intra-abdominal gonadal remnants and associated malignancy risk. In contrast, Patient 3 underwent orchidopexy and testosterone therapy prior to genetic testing. The molecular diagnosis did not directly influence initial management; it retrospectively confirmed the clinical diagnosis of PGD, although earlier genetic testing could have significantly enhanced the diagnostic process by integrating with clinical and biochemical findings, thereby enabling the multidisciplinary team to make more informed decisions regarding patient management. It would also have supported more accurate genetic counseling, helped guide future care plans, and informed discussions with the family regarding recurrence risk in future pregnancies.
Overall, these findings underscore the growing importance of early genetic testing in DSD evaluation, not only for precise diagnosis but also for anticipating long-term endocrine, surgical, and psychosocial management needs. Given the phenotypic overlap between DHX37-related DSD and mutations in other genes such as NR5A1 and MAP3K1, WES serves as a valuable tool to guide individualized care (21).
Functionally, DHX37 may influence testicular development by modulating the WNT signaling pathway, which is dosage-sensitive and essential for gonadal differentiation. Disruptions at critical developmental stages may lead to a spectrum of outcomes, from partial dysgenesis to complete regression. Zidoune et al. reported the only known homozygous DHX37 mutation (p.T477M) in a French TRS patient. Interestingly, the patient’s father—although fertile—had unilateral testicular agenesis, supporting autosomal dominant inheritance with high penetrance (22).
A review by Peng et al. identified 60 patients with DHX37-related 46,XY DSD, with p.Arg308Gln and p.Arg674Trp being the two most common variants, accounting for 36.67% and 11.67% of cases, respectively. The detection frequency of DHX37 mutations varies across DSD cohorts (0.77%–45.45%) and is particularly high in TRS and 46,XY gonadal dysgenesis subgroups (23). Notably, 46,XX female carriers appear unaffected in terms of gonadal development and fertility, while incomplete penetrance is observed in 46,XY males (23).
The management primarily involves surgical intervention and hormone replacement therapy at appropriate developmental stages. However, the long-term prognosis remains uncertain. Although the molecular mechanisms are not yet fully understood, alterations in ribosome synthesis, cell cycle regulation, NF-kB, and WNT signaling have been implicated (23).
A limitation of our study is the lack of functional validation for the novel p.Gly478Val variant. Future in-vitro or in-vivo studies are needed to determine its impact on RNA helicase activity and ribosome biogenesis. Long-term follow-up will also be essential to monitor pubertal progression and fertility outcomes, particularly in patients undergoing hormonal or surgical intervention.
In conclusion, our case series contributes to the growing evidence supporting DHX37 as a key gene implicated in 46,XY DSD. The identification of a novel variant in a Saudi patient expands the known mutational landscape and reinforces the importance of genetic testing for ambiguous genitalia, particularly in populations with high consanguinity. Ongoing research is needed to elucidate the molecular mechanisms by which DHX37 mutations impair gonadal development and to identify potential therapeutic targets.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lee PA Houk CP Ahmed SF Hughes IA . International Consensus Conference on Intersex. Consensus statement on management of intersex disorders. Pediatrics. (2006) 118:e 488–500. doi: 10.1542/peds.2006-0738, PMID: 16882788 · doi ↗ · pubmed ↗
- 2Lee PA Nordenström A Houk CP Ahmed SF Auchus R Baratz A . Global disorders of sex development update since 2006: perceptions, approach and care. Horm Res Paediatr. (2016) 85:158–80. doi: 10.1159/000442975, PMID: 26820577 · doi ↗ · pubmed ↗
- 3El Beck MD Germano CW Barros BA Andrade JG Guaragna-Filho G Paula GB . Why pediatricians need to know the disorders of sex development: experience of 709 cases in a specialized service. J Pediatr (Rio J). (2020) 96:607–13. doi: 10.1016/j.jped.2019.04.007, PMID: 31254527 PMC 9432188 · doi ↗ · pubmed ↗
- 4Bashamboo A Mc Elreavey K . Mechanism of sex determination in humans: insights from disorders of sex development. Sex Dev. (2016) 10:313–25. doi: 10.1159/000452637, PMID: 27915330 · doi ↗ · pubmed ↗
- 5Hegarty PK Mushtaq I Sebire NJ . Natural history of testicular regression syndrome and consequences for clinical management. J Pediatr Urol. (2007) 3:206–8. doi: 10.1016/j.jpurol.2006.08.007, PMID: 18947736 · doi ↗ · pubmed ↗
- 6Heksch RA Matheson MA Tishelman AC Swartz JM Jayanthi VR Diamond DA . Testicular regression syndrome: practice variation in diagnosis and management. Endocr Pract. (2019) 25:779–86. doi: 10.4158/EP-2019-0032, PMID: 31013155 · doi ↗ · pubmed ↗
- 7da Silva TE Gomes NL Lerário AM Keegan CE Nishi MY Carvalho FM . Genetic evidence of the association of DEAH-box helicase 37 defects with 46,XY gonadal dysgenesis spectrum. J Clin Endocrinol Metab. (2019) 104:5923–34. doi: 10.1210/jc.2019-00984, PMID: 31287541 · doi ↗ · pubmed ↗
- 8Mc Elreavey K Pailhoux E Bashamboo A . DHX 37 and 46,XY DSD: A new ribosomopathy? Sex Dev. (2022) 16:194–206. doi: 10.1159/000522004, PMID: 35835064 · doi ↗ · pubmed ↗