Direct whole-genome sequencing enables strain typing of unculturable Neisseria meningitidis from oropharyngeal carriage specimens
Rosa C. Coldbeck-Shackley, Andrew Lawrence, Mark McMillan, Caitlin A. Selway, Lito Papanicolas, Mark Turra, Helen Marshall, Lex E. X. Leong

TL;DR
Researchers developed a method to directly sequence the genome of Neisseria meningitidis from throat samples, even when the bacteria can't be cultured, enabling better tracking of disease-causing strains.
Contribution
The study introduces a direct whole-genome sequencing method for unculturable Neisseria meningitidis samples, enabling high-resolution strain typing.
Findings
Direct whole-genome sequencing achieved high sensitivity for genogroup and porA typing in unculturable Neisseria meningitidis samples.
Phylotyping accurately predicted clonal complexes in 93.46% of samples, showing promise for strain classification.
The proportion of Neisseria meningitidis sequences and co-occurring Neisseria species affected the accuracy of molecular typing.
Abstract
Oropharyngeal carriage of Neisseria meningitidis (N.m.) is a prerequisite for invasive meningococcal disease. As such, genomic surveillance of disease-causing carriage strains can inform targeted public health responses. However, whole-genome sequencing (WGS) from isolates is often precluded due to the high rates of culture failure for N.m. samples collected in carriage studies. This study outlines an alternative method to sequence N.m. directly from oropharyngeal specimens that enables high-resolution molecular fine typing. We performed direct probe-capture enrichment WGS (dWGS) of N.m. on oropharyngeal specimens from the ‘B part of it’ South Australian and ‘B part of it NT’ Northern Territory meningococcal carriage studies (NCT03089086 and NCT04398849). Sequences were analysed using currently available bioinformatic tools, including the characterization of genogroup, multi-locus…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
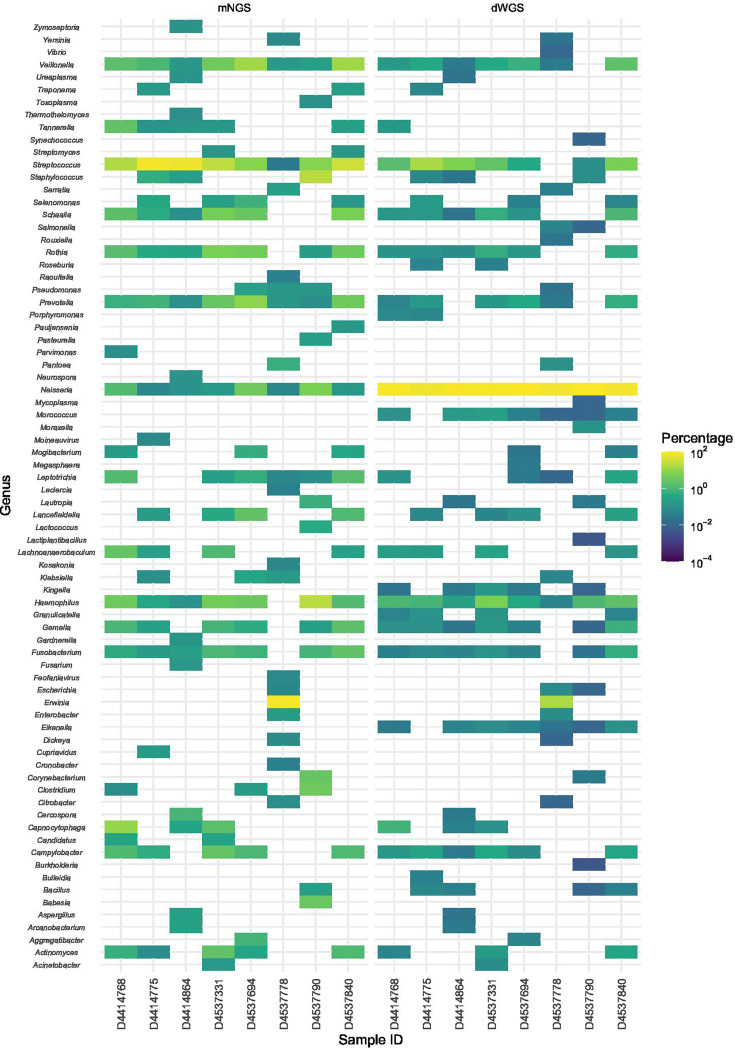 Fig. 1
Fig. 1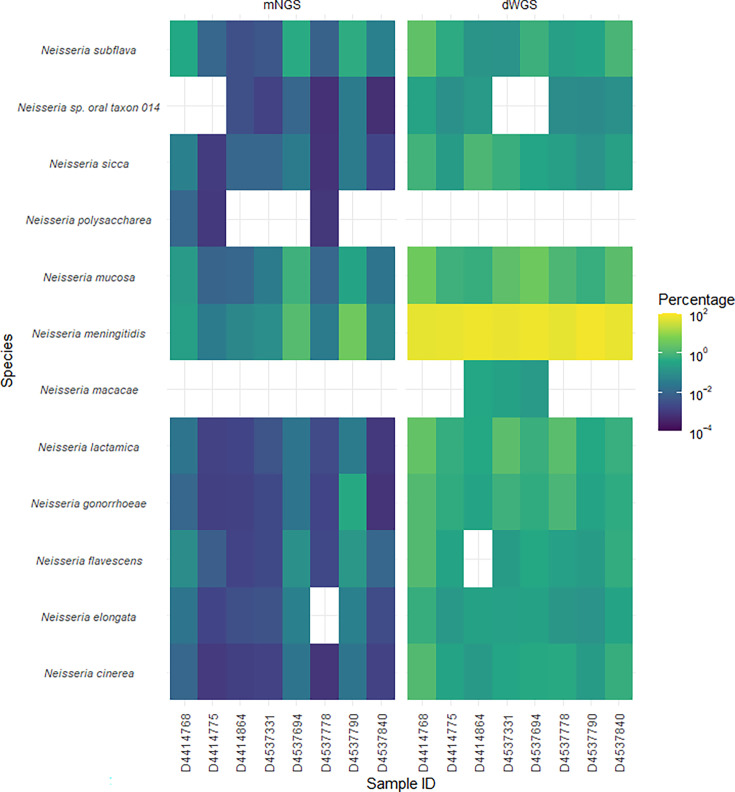 Fig. 2
Fig. 2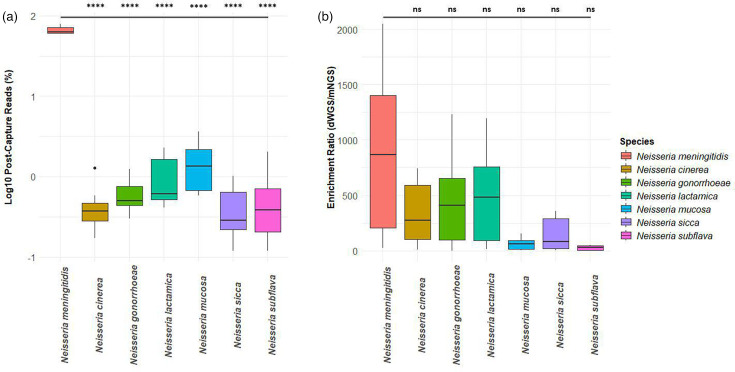 Fig. 3
Fig. 3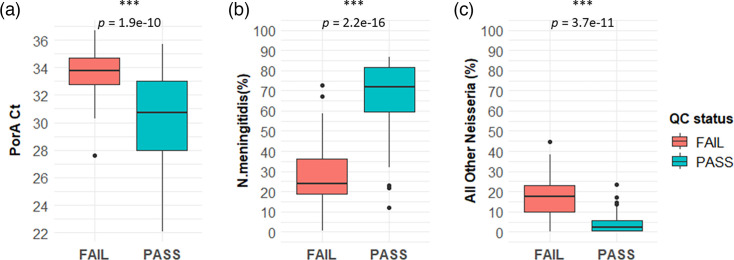 Fig. 4
Fig. 4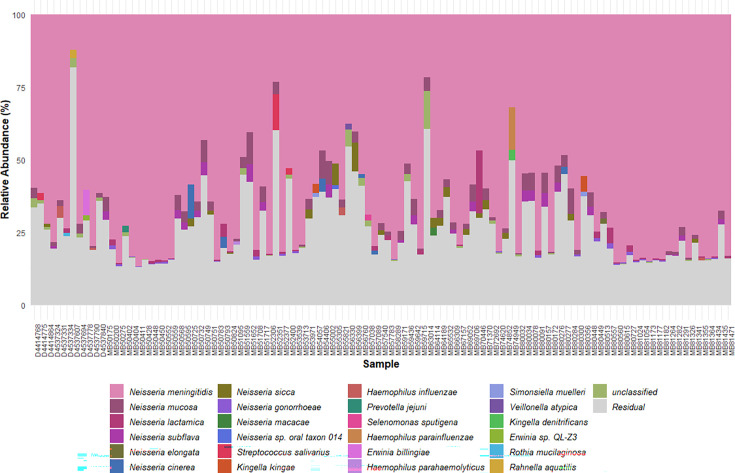 Fig. 5
Fig. 5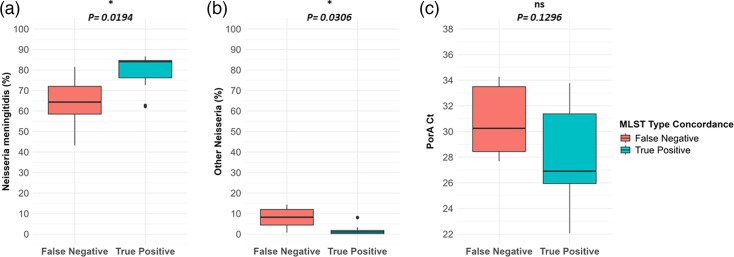 Fig. 6
Fig. 6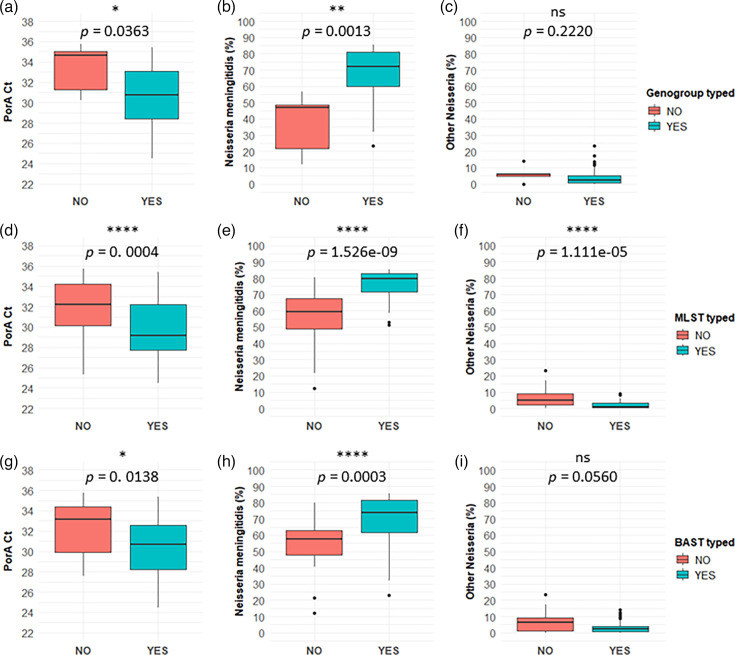 Fig. 7
Fig. 7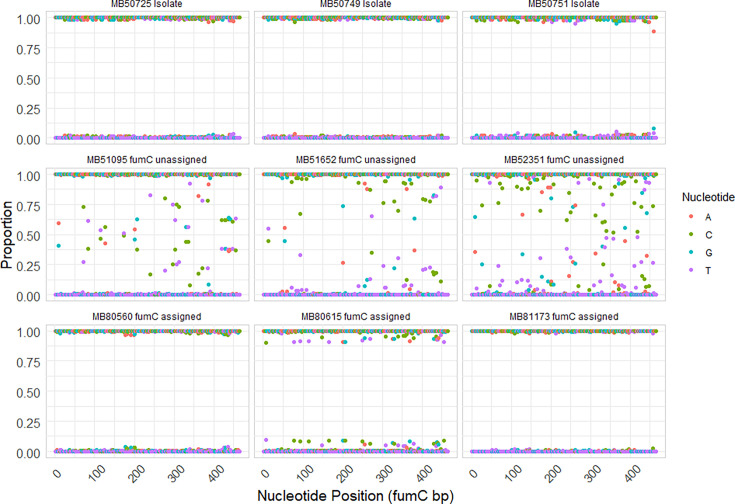 Fig. 8
Fig. 8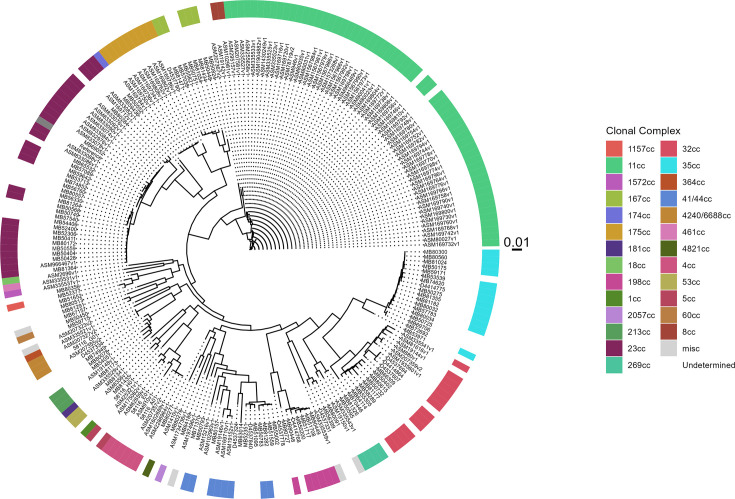 Fig. 9
Fig. 9- —http://dx.doi.org/10.13039/100020955 GlaxoSmithKline Biologicals
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Infections and Vaccines · Infective Endocarditis Diagnosis and Management · Pneumonia and Respiratory Infections
Data Summary
Raw sequence data for all samples presented in this study are available via the National Center for Biotechnology Information (NCBI) sequence read archive (BioProject: PRJNA1237918, see Table S1 for accessions). Source code is available at https://github.com/RColdbeck-Shackley/direct_WGS-of-Nmeningitidis.
Introduction
Neisseria meningitidis (N.m.) is the causative agent of invasive meningococcal disease (IMD), a potentially fatal disseminated infection that causes meningitis and septicaemia. Although the incidence of IMD in Australia is rare (0.47 per 100,000 cases in 2015), it is associated with high mortality rates in children and infants, with a second peak occurring during adolescence corresponding to increased carriage rates in these demographics [12].
Asymptomatic carriage of N.m. in the oropharynx is common in adolescence and young adulthood [3]. Asymptomatic carriers of N.m. hypervirulent strains are the most significant reservoir for new IMD cases [34]; thus, monitoring carriage in the community is informative for public health interventions.
Certain N.m. strains are known risk factors associated with an individual developing IMD [5]. Meningococci have historically been categorized by serogroup based on antibody-mediated detection of capsule antigens. In Australia, the capsule type (genogroup) is primarily determined by PCR detection of serogroup-specific capsule biosynthesis genes. However, PCR-based methods have limited resolution, cannot identify commonly carried non-groupable strains and require multiple assays to determine sub-capsular strain variation [67]. Sub-capsular typing (fine typing) enables identification of hypervirulent strains more likely to cause disease, enhances epidemiological discrimination, improves outbreak traceability compared to genogroup alone, can identify likely capsule switching events and can predict vaccine coverage [6,810].
Whole-genome sequencing (WGS) has streamlined N.m. fine typing since it enables assessment of multiple typing schemes simultaneously. This includes characterization of the porA and fetA variable regions and Neisseria multi-locus sequence typing (MLST) based on seven core-genes under stabilizing selection (abcZ, adk, aroE, fumC, gdh, pdhC and pgm) [8]. MLST sequence types (ST) can be further clustered into clonal complexes (cc) that are useful for identifying common disease-causing strains [11]. Most IMD in Australia are associated with hyperinvasive strains, such as genogroup B (MenB) cc41/44, MenY cc23 and MenW cc11 [1213]. Additionally, the Bexsero Antigen Sequence Typing (BAST) scheme classifies variants based on the fHbp, NadA, NHBA and PorA vaccine antigens and is used to predict vaccine efficacy of the MenB vaccines Bexsero and Trumenba [14].
Although WGS is useful for N.m. fine typing, it is normally limited to cultured isolates. N.m. is notoriously susceptible to temperature and other environmental stressors, often resulting in suboptimal culture rates [1516]. Recently, we evaluated the effect of the 4CMenB vaccination on the genomic epidemiology of N.m. carriage from the ‘B Part of It*’* study, an Australian cluster randomized controlled trial conducted between 2017 and 2020 in South Australia (SA) [6], and then ‘B part of it NT’, an observational study in the Northern Territory (NT) was conducted in 2021 to 2024 [17]. Despite optimized sample storage and culture conditions [1819], ~30% of samples that screened positive for N.m. by porA PCR from the SA ‘B Part of It’ study were unculturable. Sample viability was even lower for NT samples, likely due to extra transport distances and hot weather conditions in this part of Australia.
Methods for direct sequencing of pathogens from clinical specimens are now available, but challenges exist. Primary specimens often contain high concentrations of nucleic acid from the host and/or other microbes compared to the pathogen of interest. Deep sequencing and metagenomic analysis can improve sequencing depth of the pathogen of interest but are expensive and often ineffective at resolving subspecies variation from highly microbially diverse samples [20]. Alternative methods utilize PCR amplification or probe-capture enrichment of target pathogen nucleic acid from metagenomic sequencing libraries [2122]. Probe capture enrichment WGS has been successfully applied to IMD samples from sterile sites (blood, cerebrospinal fluid (CSF)) for capsular transport gene (ctrA) PCR up to a cycle threshold (Ct) of 35 [22]. However, it has not been attempted on microbiologically complex samples like the oropharyngeal specimens used in carriage studies [23].
This study aimed to determine the sensitivity of a direct probe-capture enrichment WGS (dWGS) method compared to isolating WGS for fine-typing N.m. carriage strains from oropharyngeal swabs using currently available bioinformatic tools. Further, we use dWGS to characterize unculturable N.m. collected as part of the SA and NT ‘B part of it*’* carriage studies.
Methods
Sample selection
A total of 161 PCR confirmed oropharyngeal swab specimens with porA Ct≤35 were selected for testing from the ‘B Part of It’ (n=137) and ‘B part of it NT’ (n=24) studies conducted in the Australian jurisdictions of SA and the NT, respectively. Initially, specimens were processed and Neisseria culture was attempted as previously described [24]. Following initial culture attempts, specimens were transferred to deep well storage blocks and frozen at −80 ^o^C until their use in this study. From South Australian participants, 114 samples were unable to be cultured (unculturable) [6]. For the remaining 23 SA samples, isolates were previously cultured (culturable) and sequenced by WGS, with typing results available for comparison [25]. All 24 NT samples were unculturable.
Nucleic acid extraction and porA PCR detection screening
External lysis buffer (Roche) was added to the swab liquid in a 1:5 ratio with a minimum of 200 µl final volume for extraction. Total DNA was extracted using the QIASymphony SP with QIASymphony DSP Virus/Pathogen Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions including the use of carrier RNA. DNA concentration was quantified using Quant-IT dsDNA High Sensitivity Kit (Thermo Fisher Scientific, MA, USA). Extracts were screened for the presence of N.m. by porA PCR using a previously published assay [26].
NGS library preparation, probe-capture enrichment and sequencing
Library preparation and probe-capture enrichment were performed using the Agilent SureSelect XT Low Input Target Enrichment system following the recommended protocol including modifications for sequencing of viruses and bacteria. Specifically, N.m community design probes (5,191–6,712, Agilent) were diluted 1:10 prior to use, and 2 µl of diluted probe was added to individual sample hybridization reactions. Pre- and post-capture PCR were performed for 13 cycles and 18 cycles, respectively. PCR clean-up was performed using AxyPrep MAG PCR Clean-Up Kit (Corning). DNA was quantified using Quant-IT dsDNA High Sensitivity Kit (Thermo Fisher Scientific), and library size was determined using a DNA 1 K/12 K/Hi Sensitivity Assay LabChip (Perkin Elmer). Post-capture libraries were pooled using equimolar ratios in batches of 32 including a negative control to assess for contamination and sequenced on a NextSeq 550 platform with NextSeq 500/550 Mid-Output Kit v2.5 (300 cycles) (Illumina Inc.). Index masking was performed using i8 and i10 parameters and paired-end sequencing was performed for 2×150 cycles.
Metagenomic next-generation sequencing bioinformatic processing
For metagenomic analyses, Agilent SureSelect XT pre-capture libraries were pooled in equimolar ratios and sequenced using the NextSeq 500/550 High Output Kit v2.5 (300 Cycles). Raw fastq paired-end reads were processed using an in-house metagenomic pipeline. Briefly, this pipeline performs read trimming and deduplication with fastp (v0.22.0) [27], host read removal by mapping to the human reference genome (GenBank: GCA_000001405.15 GRCh38) with bowtie2 (v2.2.5) and filtering with samtools (v1.13). Microbiome profiling was determined using the k-mer taxonomic classifier kraken2 (v2.1.2) [2830].
Direct WGS probe-enriched bioinformatic processing and typing
Paired-end raw fastq files were processed using trimmomatic (v0.39) to remove Illumina adapters and low-quality bases using a 4:20 sliding window and minimum length 30 [31]. Next, trimmed reads were aligned to the human reference genome (GenBank: GCA_000001405.15 GRCh38) using bowtie2 (v2.2.5) and filtering with samtools (v1.13). Filtered reads were then built into contiguous assemblies using the shovill pipeline with spades (v1.1.0) and their quality assessed by Seqtk (v1.3-r106) packaged as fa from the nullarbor pipeline [3233]. Assemblies were aligned to a N.m. reference (GenBank: GCA_008330805.1 ASM833080v1) using lra (v1.3.2) and filtered using samtools (flags -F 4 and -F 2048) to remove contaminant contigs [34]. Filtered assemblies were input to mlst (v2.19.0), meningotype (v0.8.5) and characterise_neisseria_capule tools for in-house typing [3537]. Filtered assemblies for samples that passed quality control (QC) assessment were also submitted to and are available via the pubMLST isolate database to confirm typing results. Genogroup was determined using the consensus of meningotype, characterise_neisseria_capule and pubMLST database results.
Sample species identification and QC assessment
Sequencing quality was assessed using Seqtk (v1.3-r106) packaged as fq from the nullarbor pipeline on host-filtered reads for sequencing read data statistics and kraken2 (v2.1.2; database: k2_pluspf downloaded on 20220607) for species identification [33]. Briefly, samples were considered as failed QC if the percentage of reads mapping to N.m was not the most abundant species identified or if there were fewer than 500,000 total N.m reads. Samples were also considered as failed QC if the second most abundant species was another Neisseria and the percentage of N.m reads was less than 40%.
Sensitivity calculations
WGS was performed on N.m isolates cultured from 23 specimens, according to Leong et al. [6]. Isolate sequences were then compared to their respective dWGS sequences that passed QC assessment, specifically looking at the genogroup, MLST, porA typing, fetA and BAST typing results. True positives (TP) were defined as concordant scheme types identified by both assays. False negatives (FN) were recorded if the scheme type was identified by WGS but was unidentified by dWGS (i.e. no exact match was identified in the pubMLST database due to missing or multiple loci being detected). Sensitivity (S) was calculated using the formula: . No false positives or true negatives were recorded precluding specificity calculations.
Phylotyping
Samples that passed QC assessment were also further used for a phylotyping approach using core SNP alignments generated using snippy (v4.6.0) with filtered reads mapping to a reference (RefSeq: GCA_008330805.1) on default settings [38]. One hundred and twenty-two high-quality N.m reference sequences were downloaded from the NCBI RefSeq database and were used as contextual samples and processed using snippy with contig settings (see Table S2 for accession numbers, available in the online Supplementary Material). A full core SNP alignment including all samples and references was then generated with snippy-core (v4.6.0) and used as input for FastTree (v2.1.11) with a gtr+gamma model [39]. The resultant tree was visualized and annotated using ggtree (v3.6.2) [40].
Statistical analyses
Data distributions were assessed using the Shapiro–Wilk test. For comparisons between multiple groups of equal size, Welch’s ANOVA with Games–Howell multiple comparisons was used. For comparison between two groups of unequal size, the Wilcoxon rank sum test was used. All statistical analyses were performed in R (v4.2.2).
Results
Enrichment of N.m. sequences from oropharyngeal samples by dWGS
To determine if dWGS enriched for N.m., species identification was performed on pre-capture metagenomic [metagenomic next-generation sequencing (mNGS)] and post-capture (dWGS) sequences from the same library preparation in parallel for a subset of the unculturable samples (n=8). Fig. 1 shows that the pre-capture libraries contained sequences from diverse commensal bacteria. Neisseria was represented in the top 20 genera by percentage in pre-capture libraries for all eight samples but was present in lower proportions compared to other genera such as Streptococcus, Haemophilus or Rothia. Following probe-capture enrichment, the percentage of Neisseria genus reads increased compared to other commensals in all post-capture samples.
The 20 highest represented genera in pre-capture metagenomic (mNGS) or post-capture enriched (dWGS) sequencing libraries from oropharyngeal samples with detectable N.m.
Next, the specificity of the enrichment process for N.m. compared to other Neisseria species was assessed. Fig. 2 demonstrates dWGS enriched for N.m. and, to a lesser extent, other Neisseria species. Although the percentage of post-capture N.m. reads was significantly greater compared to the other Neisseria (Fig. 3a), enrichment ratios were not significantly different between Neisseria species (Fig. 3b). Collectively, this demonstrates that the dWGS method enriched for N.m. sequences, but non-specific enrichment of other Neisseria species also occurred.
The percentage of reads mapping to the top ten most prevalent Neisseria species present in pre-capture metagenomic (mNGS) and post-capture enriched (dWGS) sequencing libraries.
Pre- and post-capture reads were compared for the Neisseria species that were identified in all samples (n=8) sequenced in parallel by mNGS and dWGS, respectively. (a) Log10 transformed read percentages mapping to Neisseria species in post-capture libraries. (b) Read percentage enrichment ratios for Neisseria species were calculated by the read percentages from post-capture libraries (dWGS) divided by read percentages from pre-capture libraries (mNGS) independently for each species in each sample. Statistics were performed by Welch’s ANOVA with Games–Howell multiple comparisons. Statistical significance is summarized as follows for P-value: > 0.05 [non-significant (ns)], ≤ 0.05 (), ≤ 0.01 (), ≤ 0.001 (), ≤ 0.0001 (**).
Quality control assessment of probe-capture enriched dWGS samples
Since the dWGS approach also enriched other Neisseria species, we implemented a quality control (QC) assessment that excluded samples with a high proportion of non-meningitidis reads from further analysis. The factors associated with samples that passed or failed this QC assessment were then analysed. Following dWGS, 107 out of all 161 probe-capture enriched samples passed QC assessment (66.46%; 95% CI: 58.54–73.58%). The remaining 54 samples that failed QC assessment were excluded from further analyses (see Table S1 for all QC data). Samples that passed QC had a significantly lower median porA Ct compared to those that failed (median: PASS=30.69, FAIL=33.78; Wilcoxon *P-*value=1.871e−10) (see Fig. 4a). Likewise, the percentage of N.m. reads was higher in samples that passed compared to those that failed QC assessment (median: PASS=72.02%, FAIL=24.07%; Wilcoxon P-value=2.2e−16) (see Fig. 4b). Other Neisseria species were frequently identified amongst the top three species in samples irrespective of their QC status (frequency: PASS=(97/107) 90.65%, FAIL=(46/54) 85.19%, OR=1.68; 95% CI: 0.54–5.09, Fisher’s P-value=0.3022). Despite their frequent identification, the percentage of other Neisseria species reads was lower in samples that passed QC assessment compared to those that failed (median: PASS=2.24%, FAIL=17.62%; Wilcoxon P-value=3.689e−11) (Fig. 4c). Enriched samples passing QC assessment frequently had non-Neisseria detected in the top three species including common commensal Haemophilus, Kingella, Prevotella and Streptococcus species [23] (see Fig. 5). No association was found between the presence of any non-Neisseria species in the top three with a sample passing QC assessment.
Factors associated with samples that failed or passed quality control assessment. Samples failed QC if they met the following criteria: the top species identified ≠ ‘Neisseria meningitidis’, or the number of N.m reads≤500,000, or the second species identified=other Neisseria species and the proportion of N.m reads≤40 %. (a) The porA PCR Ct of DNA extract, (b) the percentage of N.m. reads in enriched dWGS libraries identified and (c) the percentage of other Neisseria species reads in enriched dWGS libraries. Statistical comparisons were performed by the Wilcoxon rank sum test.
Top three species identified in post-capture enriched dWGS sample libraries that passed QC assessment. Samples failed QC if they met the following criteria: the top species identified ≠ ‘Neisseria meningitidis’, or the number of N.m reads≤500,000, or the second species identified=other Neisseria species and the proportion of N.m reads≤40 %. Species identified in the top three are coloured, and ‘Residual’ (grey) represents all other low-prevalence species identified in these samples.
Specificity of direct WGS compared to isolated WGS typing results
After establishing a QC assessment that reduced read contamination from other Neisseria species, the sensitivity of dWGS molecular typing was analysed compared to isolate WGS. Of the 23 culturable samples sequenced previously by isolate WGS [6], 17 passed dWGS QC. Compared to respective isolate WGS typing, the genogroup identified by dWGS was true positives for 15 samples (S=88.23%; 95% CI: 62.25–97.93%), MLST for 12 (S=70.59%; 95% CI: 44.05–88.62%), porA for 17 (S=100%; 95% CI: 77.08–100.00 %), fetA for 16 (S=94.12%; 95% CI: 69.24–99.69 %) and BAST for 15 (S=88.23%; 95% CI: 62.25–97.93%) (see Table S3 for dWGS and WGS typing matrix). Of the 5 samples that had false negative results for MLST, 23 of 35 identified alleles were concordant with the isolate-derived MLST (S=65.71%) (see Table S4 for allele matrix). Sensitivity for MLST was significantly associated with samples having a higher percentage of N.m. (Fig. 6a) and a lower percentage of other Neisseria species identified (Fig. 6b). There was no significant association with porA Ct and MLST sensitivity results (Fig. 6c). There were insufficient false negative results to statistically analyse sample factors contributing to sensitivity for the other typing schemes. Collectively, this demonstrates that dWGS can perform typing that is highly concordant with isolate WGS results. However, a lower percentage of N.m. and higher proportions of other Neisseria species in post-capture libraries reduced MLST typing sensitivity.
Factors associated with correct identification of MLST type for dWGS compared to isolate WGS for all carriage specimens with a corresponding isolate (n=17). (a) The percentage of reads in enriched dWGS libraries matching Neisseria meningitidis, (b) the percentage of reads in enriched dWGS libraries matching other Neisseria species and (c) porA PCR Ct of DNA extract. Statistics were performed by the Wilcoxon rank sum test.
Typing of unculturable samples by dWGS
After assessing the sensitivity of dWGS typing, this approach was applied to all remaining unculturable samples that passed QC assessment (n=89). PorA and fetA were characterized for 85 and 76 unculturable samples, respectively (porA, 95.50%; fetA, 85.39%). Thirty-three different porA types were identified (for a full summary of typing results, see Table S5). The most common porA types were 5–1, 10–1 (n=10, 11.23%), followed by 22, 14 (n=8, 8.99%) and 22–1, 14 (n=7, 7.87%). FetA was less diverse with 20 types being identified, and the most common were as follows: F4-1 (n=14, 15.73%), F3-9 (n=13, 14.61%) and F5-5 (n=11, 12.34%).
Eighty-four samples had genogroup identified (94.38%). Most were non-groupable (n=33, 37.08%), followed by MenY (n=21, 23.60%), MenB (n=19, 21.85%) and cnl (n=7, 7.87%). Four MenC (4.49%) were also detected in these samples.
MLST was identified for 49 samples (55.06%), including 23 different STs from 11 cc. ST-35 cc35 (n=11, 12.36%), ST-1655 cc23 (n=5, 5.62%), ST-23 cc23 and ST-823 cc198 (n=4, 4.49%) were the most abundant types identified. Additionally, this analysis identified two specimens from cc41/44, which are linked to IMD cases in Australia [13].
BAST was identified for 71 samples (79.78%) with a total of 39 types identified in the dataset. The most common BASTs were 221 and 257 (n=7, 7.87%), 228, 261, 315 and 4307 (n=4, 4.49%). These had different MenDeVAR Bexsero Reactivity scores, and BAST 315 had an exact match to the vaccine antigens, indicating MenB vaccination may provide protection against invasive disease caused by this variant. Two hundred sixty-one has no vaccine reactivity, and there were insufficient data for 221, 228, 257 and 4307 to determine reactivity based on experimental studies.
Although these numbers were small in comparison, the pattern of typing results was similar to previously analysed culture isolate WGS data from this study cohort [6].
For samples where typing results were unidentified (i.e. no exact database match due to a partial match, missing or multiple allele assignments), we postulated contributing factors were the amount and proportion of N.m. in the sample, and the presence of closely related Neisseria species, as this may interfere with typing due to sequence similarity. To interrogate this assertion porA Ct, the percentage of N.m. in sample sequence reads and the percentage of all other Neisseria species, read contaminants in ‘typed’ (YES) and ‘untyped’ (NO) samples were compared for genogroup, MLST and BAST typing schemes (see Fig. 7a–i).
Factors associated with typeability of dWGS unculturable carriage specimens (n=89). (a–c) Genogroup was compared to porA Ct, the percentage of N.m. reads or the percentage of other Neisseria species in the sample. This comparison was similarly performed for MLST (d–f) and BAST (g–i) typing results. Statistics were performed by the Wilcoxon rank sum test.
Genogroup and BAST were associated with the amount (porA Ct) and proportion of N.m. [N. meningitidis (%)] but were not significantly associated with the proportion of other Neisseria species [other Neisseria (%)] in these samples. However, for MLST, there was a significant correlation for all three factors.
Identification rates for each locus within the BAST and MLST schemes were also assessed using the pubMLST isolate search and annotation quality metrics score for all untyped samples (BAST, n=18; MLST, n=40). This analysis revealed suboptimal identification for all loci in the MLST and BAST typing schemes, especially for fumC and NHBA loci where only 35% and 56% of the untyped samples, respectively, had a known allele identified (see Table S6 for all loci).
The fumC locus was further investigated as it had the lowest identification rate. Filtered reads from three representative isolates sequenced by WGS, or three dWGS samples with or without the fumC allele assigned, were aligned to their respective closest fumC allelic sequences. Base frequencies were then assessed at each nucleotide position in the fumC locus. Fig. 8 shows that for dWGS samples with unassigned fumC alleles, the fumC locus exhibited heterogeneous base proportions. In contrast, the fumC locus from samples with assigned alleles was more similar to isolate WGS sequences, displaying relatively homogeneous nucleotide proportions, with the majority of bases being more than 80% composed of a specific nucleotide. In addition, visual inspection of read alignment samples without fumC assignment revealed variant bases were grouped on separate reads, suggesting that there were multiple fumC allele sources in the original sample.
Nucleotide proportions of filtered reads aligned to the fumC loci from representative isolates sequenced by WGS (isolate), samples sequenced by dWGS that did not have an exact fumC allele match in the pubMLST database (fumC unassigned) or samples sequenced by dWGS with an exact fumC allele match (fumC assigned).
Phylotyping as an alternative method for cc estimation
Since some samples remained without an MLST assigned due to having an incomplete MLST allelic profile characterized by fine typing, another approach was investigated. Classification at the cc level was attempted by phylogenetic analysis (phylotyping) for all samples that passed dWGS QC assessment (n=107). Fig. 9 shows that samples formed distinct clades separated by cc. All samples that had MLST assigned by fine typing were positioned on branches consistent with reference sequences in the correct cc. Samples without MLST assigned (n=44) also clustered amongst these MLST-assigned samples and references. For all samples without MLST assigned, an estimated cc (phylotype) was inferred based on the cc of other members of the clade. The phylotype was then compared against each respective partial MLST allelic profile (see Table S7 for allele profile comparison). Phylotypes were concordant with respective partial MLST profiles in 37 of 44 samples without MLST assigned by fine typing (84.01%). However, seven of these samples remained unresolved by phylotyping. Of these samples, two were branched as a single sample within a clade, precluding phylotype assignment, and five had MLST allelic profiles discordant with phylotype results. Nevertheless, phylotyping correctly assigned cc in 100 of the 107 dWGS samples (S=93.46%, 95% CI: 86.51–97.10%).
A full core SNP alignment, including all samples passing QC and representative sequences from diverse cc, was used as input for phylotyping. A tree structure was built using FastTree2 -gtr -gamma settings. Samples are coloured according to their cc derived from their previously assigned MLST type. ‘Undetermined’ samples (white) had no identified MLST or cc due to having been previously assigned incomplete or partial MLST allelic profiles. The tree scale indicates evolutionary distance.
Discussion
N.m. is notoriously sensitive to environmental and storage conditions leading to high rates of culture failure and hampering isolate WGS in carriage studies [16]. Previous studies have demonstrated ~70% sensitivity for N.m. culture from oropharyngeal or throat swabs, leaving ~30% of samples that cannot be cultured [1825]. Here, we describe a method to sequence and perform fine typing of N.m. directly from unculturable oropharyngeal carriage specimens.
Unlike blood and CSF samples from IMD cases where N.m. is the only organism present in these normally sterile sites [22], our study demonstrated oropharyngeal specimens are microbiologically complex, including the presence of nucleic acids from other Neisseria species even after probe-capture enrichment. Comparison between pre- and post-capture sequencing revealed that although nucleic acid from most commensal bacterial genera was depleted by dWGS, N.m. sequencing reads could not be further enriched from closely related Neisseria species. This is indicative of probe cross-reactivity and supports the close genetic relationships between Neisseria species that are often poorly differentiated by traditional phenotypic and genetic analyses [41]. Future studies that optimize probe-library composition, hybridization conditions or that develop bioinformatic tools better able to differentiate between related Neisseria species would greatly benefit this area of research.
This study demonstrated that the main factor impacting typing by dWGS was the proportion of N.m. sequencing reads in post-capture libraries. Unfortunately, this can only be determined post-sequencing and cannot be used as a sample selection criterion. However, based on the interquartile range of typed samples in this study, it is likely that a more stringent QC cut-off for N.m. read percentage, such as ~60–80%, would increase typing sensitivity. Although porA Ct was a strong indicator of whether samples passed or failed QC, it was only moderately associated with typeability for multi-locus schemes using existing bioinformatic typing tools, especially for genogroup or BAST. Unlike for CSF and blood specimens sequenced using a similar approach [22], this lack of a strong association between sequencing outcome and pre-screening PCR Ct likely reflects the microbiological complexity of oropharyngeal specimens. Although pre-screening based on porA Ct excludes some data loss, it is not the sole predictor of sequencing outcome in complex specimens. However, based on these results, a lower cut-off Ct for oropharyngeal specimens should be introduced to maximize sequencing efficiency. In this study, the median Ct for samples that passed QC assessment was ~Ct 30; this could be used as a guide for sample selection in future dWGS carriage studies.
MLST results were also significantly impacted by the presence of other Neisseria species, reflecting that the MLST scheme targets highly conserved loci across all Neisseria species [8]. Indeed, analysis of the fumC locus showed that these oropharyngeal samples are more likely to have multiple fumC alleles, likely sourced from other Neisseria species. Significant reductions in NHBA allele identification were also observed despite BAST not being significantly associated with the presence of other Neisseria species (P=0.05599). This may be due to the relatively small sample size in this study. Notably, a study by Muzzi et al. demonstrated that fHbp and NHBA antigens are conserved amongst many Neisseria species, whereas NadA was only found in N. cinerea, potentially explaining differences in locus identification [42]. The same study also demonstrated intra-genus variability of NHBA was akin to the level of variability amongst N. meningitidis. Together with results from our study, it suggests that BAST is likely impacted by the presence of other Neisseria, but possibly to a lesser extent compared to the highly conserved MLST scheme. Despite this, the ability of dWGS to determine BAST adds important information regarding the predicted MenB vaccine reactivity in unculturable carriage samples not otherwise able to be captured by isolate WGS. Unlike the MLST and BAST typing alleles, capsule expression was historically considered a unique feature of N. meningitidis, as this is the key virulence determinant for this pathogenic Neisseria species [5]. However, recent studies have identified homologues of capsule expression genes in multiple non-pathogenic Neisseria species [43]. Despite this, we found no evidence that these homologues impacted genogroup results that could be identified in ~94% of unculturable samples. Sequence similarity may also affect direct sequencing of other Neisseria species, such as Neisseria gonorrhoeae, in microbially complex specimens like the urogenital tract, where other Neisseria species can also colonize or cause infections [4445]. Future studies should be interpreted accordingly. Aside from multi-locus typing schemes, the assessment of the porA variable region produced the most consistent typing results, likely owing to it being uniquely expressed by N.m. despite pseudogenes existing in other Neisseria [46]. This may be a useful typing scheme for dWGS carriage studies in the future.
Despite the influence of other Neisseria contaminant reads on N.m. fine typing results, we observed a strong phylogenetic signal that categorized samples at the level of the clonal complex. This phylotyping approach may prove to be a useful secondary analysis where fine-typing results are not resolved by other approaches. This is likely due to the filtering of sequences from other Neisseria species during the time of read alignment to the N.m. reference genome and the consideration of additional genomic regions aside from highly conserved typing loci.
This study has limitations. Aside from the small sample size that limited the statistical significance of some analyses, since multiple N.m. strains can co-habit the oropharyngeal and nasopharyngeal sites [316], it could not be ruled out that this also impacted typing results. Despite capturing a wide variety of strains, not all genogroups were represented in the data, as many commensal N.m. strains are unencapsulated; therefore, we do not know if this method is biassed towards typing of more commonly sequenced genogroups.
Collectively, our data demonstrate that dWGS is a viable method to analyse unculturable N.m. oropharyngeal specimens. Since the meningococcal types identified in these unculturable specimens were consistent with published results and fewer in number than those from previously sequenced cultured isolates, this study does not alter any conclusions from the ‘B part of it’ genomic analysis study [25]. However, this approach enabled the characterization of meningococci to a level equivalent to clonal complex for ~93.46% of unculturable samples that were previously unable to be sequenced using standard WGS. This adds to work by others using similar methods, enabling the sequencing of uncultured IMD clinical specimens [22]. However, due to the presence of other Neisseria species commonly present in oropharyngeal sites, this method is most suited to N.m. specific typing schemes such as genogroup or the porA gene and, additionally, for whole-genome phylogenetic approaches to typing.
Supplementary material
10.1099/mgen.0.001464Uncited Supplementary Material 1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Archer BN Chiu CK Jayasinghe SH Richmond PC Mc Vernon J et al Epidemiology of invasive meningococcal B disease in Australia, 1999-2015: priority populations for vaccination Med J Aust 201720738238710.5694/mja 16.0134029092704 · doi ↗ · pubmed ↗
- 2Marshall HS Lally N Flood L Phillips P First statewide meningococcal B vaccine program in infants, children and adolescents: evidence for implementation in South Australia Med J Aust 2020212899310.5694/mja 2.5048131909501 · doi ↗ · pubmed ↗
- 3Caugant DA Maiden MCJ Meningococcal carriage and disease--population biology and evolution Vaccine 200927 Suppl 2B 647010.1016/j.vaccine.2009.04.06119464092 PMC 2719693 · doi ↗ · pubmed ↗
- 4Christensen H May M Bowen L Hickman M Trotter CL Meningococcal carriage by age: a systematic review and meta-analysis Lancet Infect Dis 20101085386110.1016/S 1473-3099(10)70251-621075057 · doi ↗ · pubmed ↗
- 5Rouphael NG Stephens DS Neisseria meningitidis: biology, microbiology, and epidemiology Methods Mol Biol Clifton NJ 201279912010.1007/978-1-61779-346-2_1PMC 434942221993636 · doi ↗ · pubmed ↗
- 6Leong LEX Coldbeck-Shackley RC Mc Millan M Bratcher HB Turra M et al The genomic epidemiology of Neisseria meningitidis carriage from a randomised controlled trial of 4C Men B vaccination in an asymptomatic adolescent population The Lancet Regional Health - Western Pacific 20244310096610.1016/j.lanwpc.2023.10096638169944 PMC 10758868 · doi ↗ · pubmed ↗
- 7Dolan-Livengood JM Miller YK Martin LE Urwin R Stephens DS Genetic basis for nongroupable Neisseria meningitidis J Infect Dis 20031871616162810.1086/37474012721942 · doi ↗ · pubmed ↗
- 8Jolley KA Brehony C Maiden MCJ Molecular typing of meningococci: recommendations for target choice and nomenclature FEMS Microbiol Rev 200731899610.1111/j.1574-6976.2006.00057.x 17168996 · doi ↗ · pubmed ↗