Synthesis and Conformational Analysis of Pyran Interhalide Analogues of Galactose, Mannose, Talose, and Glucose
Olivier Lessard, Mathilde Grosset‐Magagne, Pierangelo Metrangolo, Denis Giguère

TL;DR
This paper describes the synthesis and structural analysis of halogen-containing sugar analogs, revealing unique halogen bonding interactions.
Contribution
A new halogeno-divergent strategy for synthesizing pyran interhalide analogs and the discovery of the shortest F⋅⋅⋅I halogen bond.
Findings
A robust halogeno-divergent strategy was developed for synthesizing pyran interhalide analogs of d-galactose, d-mannose, d-talose, and d-glucose.
Solid-state conformational analysis revealed differences in intra-annular torsion angles among the interhalide analogs.
The shortest F⋅⋅⋅I halogen bonding was observed involving Csp3-bound iodine and fluorine atoms.
Abstract
There is growing interest in the preparation of multivicinal interhalide molecules. Herein, we described the synthesis of several pyran interhalide analogues of d‐galactose, d‐mannose, d‐talose, and d‐glucose. A robust and simple halogeno‐divergent strategy provided pyran‐bearing multivicinal interhalide stereocenters. However, a different strategy was implemented for the synthesis of glucose interhalides. The solid‐state conformational analysis of some interhalide analogues showed differences in the deviation of the intra‐annular torsion angles. Moreover, we observed the shortest F⋅⋅⋅I halogen bonding (XB) involving Csp3‐bound I and F atoms. The synthesis and conformational analysis of pyran interhalides unveiled the shortest F⋅⋅⋅I halogen bonding involving Csp3‐bound I and F atoms.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
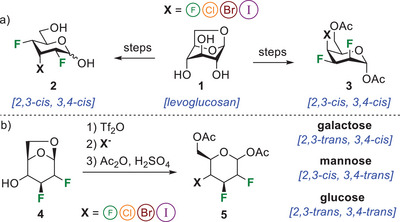 Figure 1
Figure 1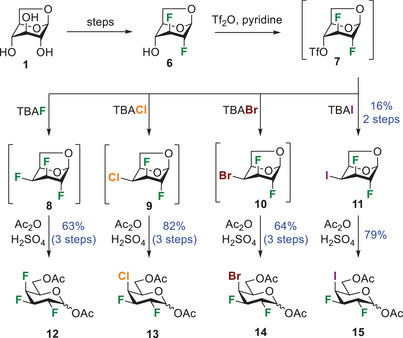 Scheme 1
Scheme 1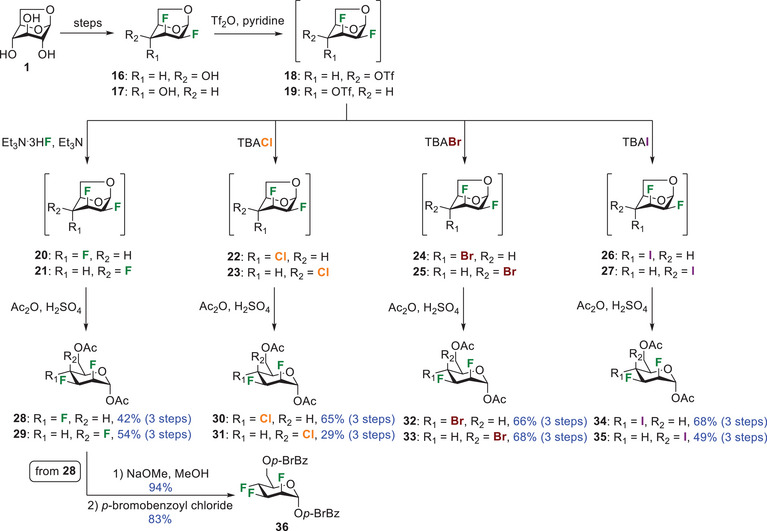 Scheme 2
Scheme 2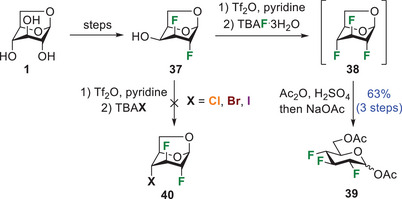 Scheme 3
Scheme 3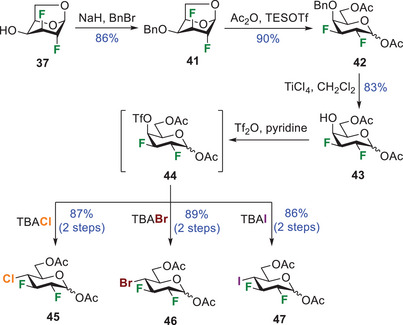 Scheme 4
Scheme 4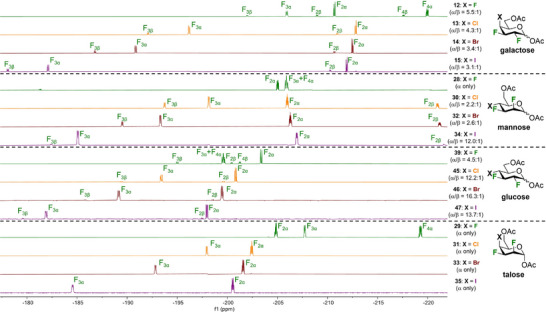 Figure 2
Figure 2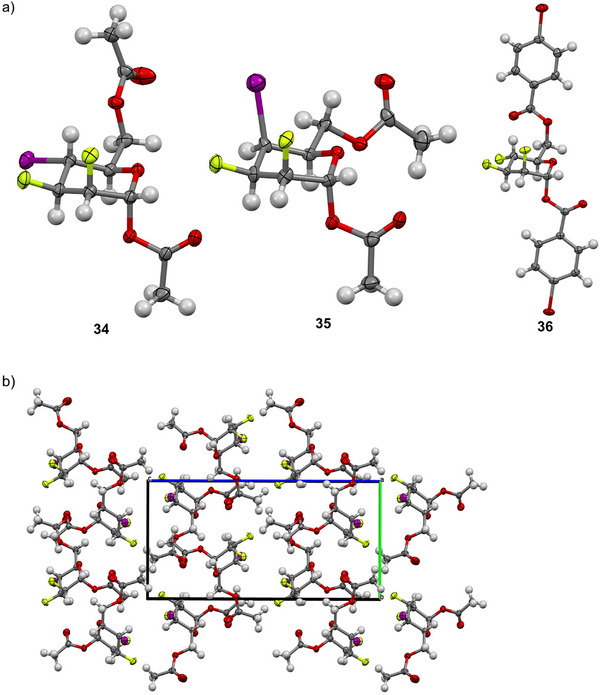 Figure 3
Figure 3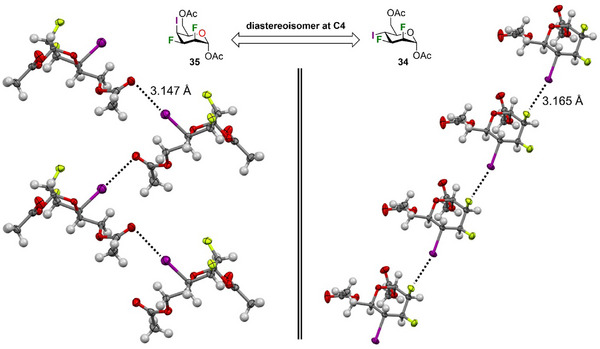 Figure 4
Figure 4|
| ||||
|---|---|---|---|---|
| Entry | Compounds | α1 (°) | α2 (°) | α1 + α2 (°) |
| 1 |
| 1.30 | 0.81 | 2.11 |
| 2 |
| 7.06 | 11.53 | 18.59 |
| 3 |
| 1.00 | 5.15 | 6.15 |
| 34 | 35 | 36 | |
|---|---|---|---|
|
| 0.559 | 0.523 | 0.529 |
|
| 5.235 | 7.248 | 1.609 |
|
| 263.056 | 307.711 | 149.255 |
- —Fondazione Cariplo10.13039/501100002803
- —Natural Sciences and Engineering Research Council of Canada10.13039/501100000038
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCarbohydrate Chemistry and Synthesis · Seaweed-derived Bioactive Compounds · Marine Sponges and Natural Products
Introduction
1
Polyfluorinated carbohydrates have great biological potential, with many examples reported thus far.^[^ 1 ^]^ Pyran interhalide analogues of carbohydrates are scarcer, and only a few examples are reported as useful tools in glycobiology.^[^ 2 ^]^ The lack of efficient synthesis of sugar interhalides might explain the limited number of biological investigations of these interesting organohalogens. Only a handful of reports shows the study of synthetic^[^ 3 ^]^ and natural products^[^ 4 ^]^ having multivicinal interhalide stereocenters. Nevertheless, the incorporation of halogens into small molecules can improve cellular uptake and enhance membrane binding and permeation.^[^ 5 ^]^ Related to biological systems, the increase in binding affinity arises from the rich palette of noncovalent interactions given by halogen atoms, including halogen bonding (XB), that is, the interaction involving halogen atoms as electrophilic sites.^[^ 6 ^]^ XB was also successfully used for the activation of glycosyl halides.^[^ 7 ^]^
From our end, we developed synthetic routes to multivicinal organofluorines to unveil some of their unique properties, such as the lipophilicity, conformation, and biological activities.^[^ 8 ^]^ Moreover, we developed a halo‐divergent synthetic route of pyran interhalide analogues of d‐allose 2 from levoglucosan 1 (Figure 1a).^[^ 9 ^]^ Allopyranose interhalides 2 (incorporating the 2,3‐cis, 3,4‐cis relationship for the halogens) was the starting point to complex trihalo‐ and tetrahalo‐hexanetriols. Also, halogenated analogues of Pitolisant were prepared, highlighting an approach to increase the molecular dipole of piperidines. Recently, we reported the synthesis of pyran interhalide analogues of d‐talopyranose 3 that also integrates the 2,3‐cis, 3,4‐cis relationship for the halogens (Figure 1a).^[^ 10 ^]^ Talose interhalides 3 showed distinct solid‐state conformations because of 1,3‐diaxial repulsion between fluorine at C2 and halogen atoms. Additionally, we reported the first application of XB in the context of solid‐state ordering of pyran interhalides. Lastly, the group of Linclau reported the synthesis and lipophilicity evaluations of a series of dideoxygenated chloro‐fluorosugars.^[^ 11 ^]^ It was shown that the log P increases by an average of 1.37 log P units for the deoxychlorination products as compared to 0.83 log P for the corresponding deoxyfluorination products.
*Synthesis of trihalogenated pyrans: a) Chiron approach to pyran interhalide analogues of allopyranoses 2 and talopyranoses 3;[
9 , 10
] b) Synthetic strategy to multivicinal interhalide analogues of d‐galactose, d‐mannose, and d‐glucose (this work).*
We wish to extend our approach to the synthesis of three sets of pyran interhalide analogues from 1,6‐anhydro‐2,3‐dideoxy‐2,3‐difluoro‐β‐d‐hexopyranoses 4 (Figure 1b). A robust and simple strategy involving a halogeno‐divergent route provided d‐galactose (2,3‐trans, 3,4‐cis relationship for the halogens), d‐mannose (2,3‐cis, 3,4‐trans relationship for the halogens), and d‐glucose (2,3‐trans, 3,4‐trans relationship for the halogens). The solid‐state conformational analysis and comparison of ^19^F resonances of some analogues add more data to the field of polyhalogenated molecules.
Results and discussions
2
Our recent discovery that the nature and the stereochemistry of halogen atoms can have a strong impact on conformation and lipophilicity motivated us to investigate original pyran interhalides.^[^ 9, 10 ^]^ We first targeted galactopyranose interhalide analogues using an halo‐divergent synthetic route starting from known 1,6‐anhydro‐2,3‐dideoxy‐2,3‐difluoro‐β‐d‐glucopyranose 6,^[^ 8, 12 ^]^ readily accessible from levoglucosan 1 (Scheme 1). Compound 6 was activated as a triflate, providing intermediate 7, which was directly treated with a nucleophilic halogen source, providing the desired halogen substitution at C4 with inversion of configuration. Intermediates 8 − 10 were advanced to the next step without further purification, except for compound 11 because of the formation of side products after the iodination step. The halogenated 8 − 11 were treated under acetolysis conditions, providing galactopyranose analogues 12 − 15 in modest to good yields. The novel analogues integrate a 2,3‐trans, 3,4‐cis relationship for the halogens.
Synthesis of halogenated galactopyranose analogues 12 − 15 that integrate a 2,3‐trans, 3,4‐cis relationship for the halogens.
Next, we focused on the synthesis of mannopyranose interhalide analogues using the same strategy. As a point of comparison, we also report here the synthesis of talopyranose analogues.^[^ 10 ^]^ Starting from known difluorinated analogues 16 or 17,^[^ 8 ^a]^ we were able to generate triflate intermediates 18 and 19 that were subjected to nucleophilic halogenation with inversion of configuration (Scheme 2). Halogenated mannopyranoses 20, 22, 24, and 26, along with talopyranoses 21, 23, 25, and 27, were treated under acetolysis conditions, affording halogenated pyran 28 − 35. The installation of an equatorial chlorine and iodine proceeded in higher yield than the corresponding axial halogens (30: 65% vs. 31: 29% and 34: 68% vs. 35: 49%). As for the bromine, the yields were similar for both isomers (32: 66% vs. 33: 68%).
*Synthesis of halogenated mannopyranose analogues 28, 30, 32, and 34 and talopyranose analogues 29, 31, 33, and 35 that integrate a 2,3‐cis, 3,4‐trans [
10
] and a 2,3‐cis, 3,4‐cis relationship for the halogens, respectively.*
Our next task involved the preparation of halogenated glucose analogues using the same strategy as before. Thus, known 1,6‐anhydro‐2,3‐dideoxy‐2,3‐difluoro‐β‐d‐galactopyranose 37 ^[^ 8 ^a,^ 12 ^]^ was fluorinated at C4 using tetrabutylammonium fluoride (TBAF) via a triflate intermediate (Scheme 3). Acetolysis of 38 under acidic conditions provided trifluoroglucose analogue 39. Despite many efforts, this strategy failed to produce interhalides 40. In this instance, we observed formation of side products probably arising from an oxiranium‐like intermediate.^[^ 8 ^e]^ Consequently, we were compelled to use a different synthetic route to access pyran interhalide analogues of glucose.
Synthesis of trifluorinated glucose analogue 39 and failed attempt to prepare glucopyranose interhalides 40.
We rationalized that it would be easier to install a halogen equatorially on a pyran core via an S_N_2 reaction.^[^ 13 ^]^ Therefore, we cleaved the 1,6‐anhydro bridge, allowing the chair interconversion (^1^ C 4 to ^4^ C 1) prior to the nucleophilic halogenation steps. In this way, we first protected the alcohol 37 as benzyl ether in 86% yield (Scheme 4). Then, triethylsilyl triflate‐catalyzed acetolysis provided the desired diacetylated 42 in 90% yield, and a titanium tetrachloride‐mediated benzyl deprotection gave the corresponding free hydroxyl group at C4.^[^ 14 ^]^ We used the same nucleophilic substitution strategy as before. Thus, activation of the hydroxyl group 43 as triflate yielded intermediate 44 that was directly subjected to tetrabutylammonium chloride, bromide, or iodide, providing compounds 45, 46, and 47, respectively. It is worth noting that in this case, the deoxyhalogenations proceeded in high yields over 2 steps (86 – 89%).
Synthesis of glucopyranose interhalide analogues 45 – 47 that integrate a 2,3‐trans, 3,4‐trans relationship for the halogens.
With a set of pyran interhalide analogues of galactose, mannose, and glucose, we proceeded to compare their ^19^F resonance (^19^F NMR; 470 MHz, CDCl_3_) with talose interhalides (Figure 2).^[^ 10 ^]^ Firstly, all analogues adopt standard ^4^ C 1‐like conformations. The ^19^F resonance of F3 expectedly occurs at a lower field than F2 for every trihalogenated pyran analogue. In the case of the glucose analogues, both ^19^F signals shift toward lower fields as the size of the halogen atom at the C4 position increases, from −203.39 to −197.96 ppm for F2 and from −199.69 to −181.94 ppm for F3. The same trend is observed for the talose analogue with shifts increasing from − 205.46 to **− **200.55 ppm for F2 and from **− **208.33 to **− **184.56 ppm for F3. In both series, the shift variation is more important for the F3 signal than the F2 signal. A similar trend is observed for the galactose analogues, except for the F2 signal of trifluorinated 12, which is at a higher shift than the other analogues. The mannose series follows the general trend for the F3 signals, but the F2 signals surprisingly shift upfield with the increasing size of the halogen at the C4 position. The downfield shift of the F2 and F3 signals in the same series could be explained by steric deshielding.^[^ 15 ^]^ Due to the overlap of the van der Waals radii, it is hypothesized that the van der Waals forces of the halogen at the C4 position could restrict the electron motions of the fluorine atoms and therefore artificially lower their electron density. Lastly, the supporting information (Figure S1−S3) reports the ^19^F NMR predictions of acetylated pyran interhalides. Some NMR prediction tool don't consider the nature of the halogen at C4 for correctly predicting the chemical shift of fluorine atoms. For example, one of the biggest differences is the F4 signal of trifluorinated talose 29 (Δδ = −45.98 ppm). Consequently, the NMR predictions of such compounds remain very challenging, and this study adds more data to the field of NMR spectroscopy.
Direct comparison of 19F resonances (19F NMR; 470 MHz, CDCl3) of halogenated galactose analogues 12 − 15, mannose analogues 28, 30, 32, and 34, glucose analogues 39, 45 − 47, and talose analogues 29, 31, 33, and 35.
The absolute configuration of iodides 34 ^[^ 16 ^]^ and 35 ^[^ 17 ^]^ and compound 36 ^[^ 18 ^]^ were unambiguously confirmed by single‐crystal diffraction analysis. Trifluorinated analogue 28 was not crystalline; therefore we were compelled to remove the acetyl protecting groups and generate the corresponding p‐bromobenzoate derivative 36 to obtain suitable crystalline material, as previously described (Scheme 2).^[^ 8 ^a]^ Our interest in the conformation of organohalogens motivated us to compare the solid‐state conformation of halogenated pyrans 34 and 36 ^8a^ with talose interhalide 35 (Figure 3a).^[^ 10 ^]^ Compounds 34 and 35 only differ by the stereochemistry of the iodine at C4. Similarly to its analogue 36, compounds 34 and 35 adopt a standard ^4^ C 1‐like conformation in the solid state. However, as for the C5 − C6 rotamer, compound 35 exhibits a gt conformation as opposed to a gg conformation for analogues 34 and 36. For standard ^4^ C 1‐like conformation, an equatorial substituent at C4 generally leads to a gg or a gt conformation, with the tg orientation forbidden.^[^ 19 ^]^ Figure 3b shows the extended packing arrangement of mannopyranose interhalide analogues 34.
a) X‐ray analysis of compound 34 − 36; b) extended packing arrangement (view along the a‐axis) of compound 34. Oak Ridge Thermal‐Ellipsoid Plot (ORTEP) diagram showing 50% thermal ellipsoid probability: carbon (gray), oxygen (light red), fluorine (green), bromine (dark red), iodine (purple), and hydrogen (white).
We recently reported that there is a strong deviation in the intra‐annular torsion angles for compound 35 arising from the repulsion of the axial fluorine at C2 and the axial iodine at C4.^[^ 10 ^]^ Compounds 34 and 36 have an equatorial halogen at C4; thus there is a reduced 1,3‐diaxial repulsion with C2─F, leading to a limited deviation from parallel alignment as shown in Table 1. The Newman projection of halogenated analogues shows deviations from parallel alignment for the C2─F and C4 substituents of 2.11° for 34 (R_1_ = H), 6.15° for 36 (R_1_ = H), and 18.59° for 35 (R_1_ = I). Thus, the stereochemistry of one halogen (the C4 iodine) can account for a difference in the deviation in the intra‐annular torsion angles of 16.48° (34 vs. 35). The greater deviation of 36 as compared to 34 is possibly due to the large p‐bromobenzoyl group, resulting in a gg conformation. As a comparison with natural carbohydrates, the deviations are 3.50° and 7.12° for the gt and the gg rotamers of native α‐d‐mannopyranose, respectively.^[^ 20 ^]^ For native α‐d‐talopyranose, the deviation is 5.92°.^[^ 21 ^]^
**Table 1: 1,3‐Diaxial repulsion between CF2 and CH4 or CX4 bonds for compound 34 − 36.[
a
]**
We also assessed the Cremer–Pople ring puckering parameters (Table 2).^[^ 22 ^]^ These parameters take the form of a spherical polar set, Q, θ, and φ, for pyranoid rings.^[^ 23 ^]^ A smaller puckering amplitude (Q) values indicate a flattened ring. For an ideal cyclohexane chair, the puckering amplitude is 0.63 Å.^[^ 22 ^]^ The azimuthal angle (θ) represents the distortion of pyranose rings (θ = 0° is a perfect ^4^ C 1 chair, and θ = 180° represents the ^1^ C 4 chair). Expectedly, the distortion of the chair conformation of compound 35 is more important than compound 34. In addition, the nature of the distortion is indicated by the meridian angle (φ). The distortion of the iodinated mannopyranose 34 is in a direction toward a ^4^ H 5 conformation (φ ≈ 270°). The trifluorinated mannopyranose analogue 36 is distorted toward a ^2^ H 3 conformation (φ ≈ 150°), and the iodinated talopyranose analogue 35 is distorted toward an E 5 conformation (φ ≈ 300°). Once more, this set of data exemplifies the important conformational difference exerted by the stereochemistry of one iodine atom at C4 (34 vs. 35).
Some of us recently reported the first example of XB stabilizing the crystal lattice of pyran interhalides.^[^ 10 ^]^ In particular, the iodinated talose interhalide 35 shows a very directional I⋅⋅⋅O contact involving the carbonyl oxygen atom of the acetate at C6 (IC⋅⋅⋅O distance = 3.147 Å; C‐I⋅⋅⋅O angle = 179.70°) of a nearby molecule (Figure 4). The observed distance roughly corresponds to a 10% reduction of the sum of the van der Waals radii for oxygen and iodine, that is, 1.52 and 1.98 Å,^[^ 24 ^]^ respectively, which is coherent with a medium‐strength XB. Similarly to other systems involving carbonyl oxygens, the iodine approach occurs in an out‐of‐plane mode (C = O⋅⋅⋅I angle = 104.23°).^[^ 25 ^]^ Interestingly, mannose interhalide 34, instead, showed a completely different XB pattern. In particular, the C = O⋅⋅⋅I XB observed in 35 is replaced by a C‐F⋅⋅⋅I XB involving iodine and the fluorine atom at C3 (Figure 4). The F⋅⋅⋅I distance is 3.165 Å, which roughly corresponds to an 8% reduction of the sum of vdW radii for fluorine and iodine, that is, 1.45 and 1.98 Å,^[^ 24 ^]^ respectively, and the C‐I⋅⋅⋅F angle is 167.17°. The observed F⋅⋅⋅I XB is remarkably short, and, to the best of our knowledge, it is the shortest of its type, that is, involving Csp^3^‐bound I and F atoms, reported in the Cambridge Structural Database (CSD version 5.46, Update 1‐Feb 2025, 1 341 400 entries).^[^ 26 ^]^ Only FUMFUS shows a similar XB, although less directional.^[^ 27 ^]^ Whether or not organic fluorine may act as a hydrogen‐bond acceptor has been a controversial issue for more than four decades.^[^ 28 ^]^ The results reported in this article fuel the debate, clearly demonstrating that F⋅⋅⋅I interactions stabilize the crystal packing of mannose interhalides.
Packing arrangement of compound 34 and 35 showing XB interactions. ORTEP diagram showing 50% thermal ellipsoid probability: carbon (gray), oxygen (light red), fluorine (green), iodine (purple), and hydrogen (white).
Conclusions
3
We described the synthesis of 4 halogenated pyran analogues of d‐galactose, d‐mannose, d‐talose, and d‐glucose. The synthetic strategy proceeded according to plan for all analogues except for glucopyranose interhalides. In this case, cleavage of the 1,6‐anhydro bridge preceded the deoxyhalogenation step in order to avoid formation of by‐products. Direct comparison of ^19^F resonances of all halogenated analogues showed interesting trends for the chemical shifts. All analogues adopt standard ^4^ C 1‐like conformations, and the ^19^F resonance of F3 occurs at a lower field than F2 for all pyran interhalide analogues. Unexpectedly, the F2 signals of mannose shift upfield with the increasing size of the halogen at the C4 position. In the solid state, mannose interhalide 34 and talose interhalide 35 showed important conformational differences exerted by the stereochemistry of the iodine atom at C4. The Newman projections of halogenated analogues showed difference in the deviation in the intra‐annular torsion angles of 16.48°, and the Cremer–Pople ring puckering parameters showed differences in the distortion of the chair conformations. Furthermore, interhalides 34 and 35 show very different halogen bonding patterns stabilizing their crystal structures. In particular, the mannose derivative shows an unprecedented F⋅⋅⋅I halogen bonding, the shortest F⋅⋅⋅I halogen bonding involving Csp^3^‐bound I and F atoms. Finally, these findings shed more light on the intriguing nature of pyran interhalides for application in drug discovery, materials science, and other fields.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1a) Y. Zhou , J. Wang , Z. Gu , S. Wang , W. Zhu , J. L. Aceña , V. A. Soloshonok , K. Izawa , H. Liu , Chem. Rev. 2016, 116, 422–518;26756377 10.1021/acs.chemrev.5b 00392 · doi ↗ · pubmed ↗
- 2O. Lessard , D. Lainé , D. Giguère , Eur. J. Org. Chem. 2024, 27, e 202400120.
- 3Y. Tan , S. Luo , D. Li , N. Zhang , S. Jia , Y. Liu , W. Qin , C. E. Song , H. Yan , J. Am. Chem. Soc. 2017, 139, 6431.28415839 10.1021/jacs.7b 02076 · doi ↗ · pubmed ↗
- 4a) C. Bucher , C. ; R. M. Deans , N. Z. Burns , J. Am. Chem. Soc. 2015, 137, 12784;26394844 10.1021/jacs.5b 08398 PMC 4634703 · doi ↗ · pubmed ↗
- 5a) R. Cao , J. Li , Q. Liao , M. Shao , Q. Zhang , Y. Zhang , T. Cai , W. Zhu , Z. Xu , J. Med. Chem. 2025, 10.1021/acs.jmedchem.5c 00944;40335461 · doi ↗ · pubmed ↗
- 6a) G. Cavallo , P. Metrangolo , R. Milani , T. Pilati , A. Priimagi , G. Resnati , G. Terraneo , Chem. Rev. 2016, 116, 2478–2601;26812185 10.1021/acs.chemrev.5b 00484 PMC 4768247 · doi ↗ · pubmed ↗
- 7R. Castelli , S. Schindler , S. M. Walter , F. Kniep , H. S. Overkleeft , G. A. Van der Marel , S. M. Huber , J. D. C. Codée , Chem. Asian J. 2014, 9, 2095.24962953 10.1002/asia.201402259 · doi ↗ · pubmed ↗
- 8a) V. Denavit , D. Lainé , J. St‐Gelais , P. A. Johnson , D. Giguère , Nat. Commun. 2018, 9, 4721;30413697 10.1038/s 41467-018-06901-y PMC 6226540 · doi ↗ · pubmed ↗