Metastatic Pheochromocytoma in a Patient With Li-Fraumeni Syndrome
Sotiris Loizidis, Christiana Matthaiou, Efrosini Iacovou, Karel Pacak, Ashley Grossman

TL;DR
A patient with Li-Fraumeni syndrome developed metastatic pheochromocytoma, a rare occurrence in this genetic condition.
Contribution
This case highlights the unusual manifestation of metastatic pheochromocytoma in a TP53-mutated individual.
Findings
A TP53 pathogenic variant carrier developed metastatic pheochromocytoma.
Pheochromocytoma is a rare manifestation of Li-Fraumeni syndrome.
The case contributes to understanding the genomic landscape of PCCs/PGLs in TP53-mutated individuals.
Abstract
Li-Fraumeni syndrome (LFS) is a rare cancer predisposition syndrome caused by genomic alterations in the tumor protein p53 (TP53) gene. The lifetime risk of developing cancer is very high, and carriers of germline TP53 pathogenic variants must be closely monitored starting from a young age. LFS is particularly associated with specific tumors, such as breast cancer, soft tissue and bone sarcomas, primary central nervous system cancers, acute leukemia, and adrenocortical carcinoma. Despite its association with a broad spectrum of malignancies, pheochromocytoma/paraganglioma (PCC/PGL) is an unusual manifestation of LFS and has only rarely been reported. Here, we present a case of a 57-year-old female patient who is a carrier of a deleterious germline TP53 pathogenic variant and developed a PCC; several years later, she had lung and bone lesions compatible with metastatic PCC. We also…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
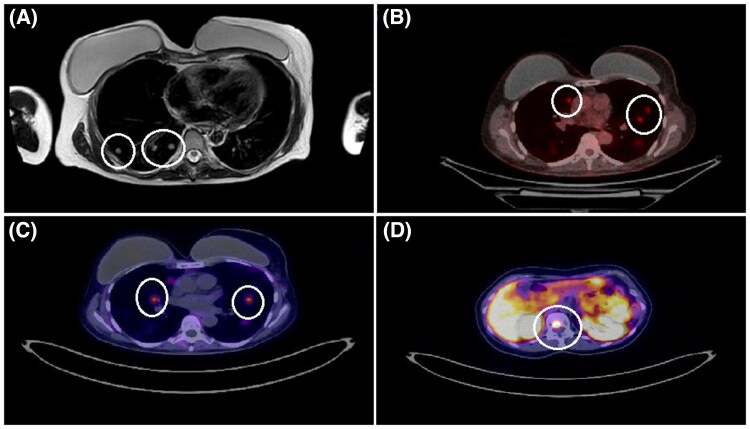 Figure 1
Figure 1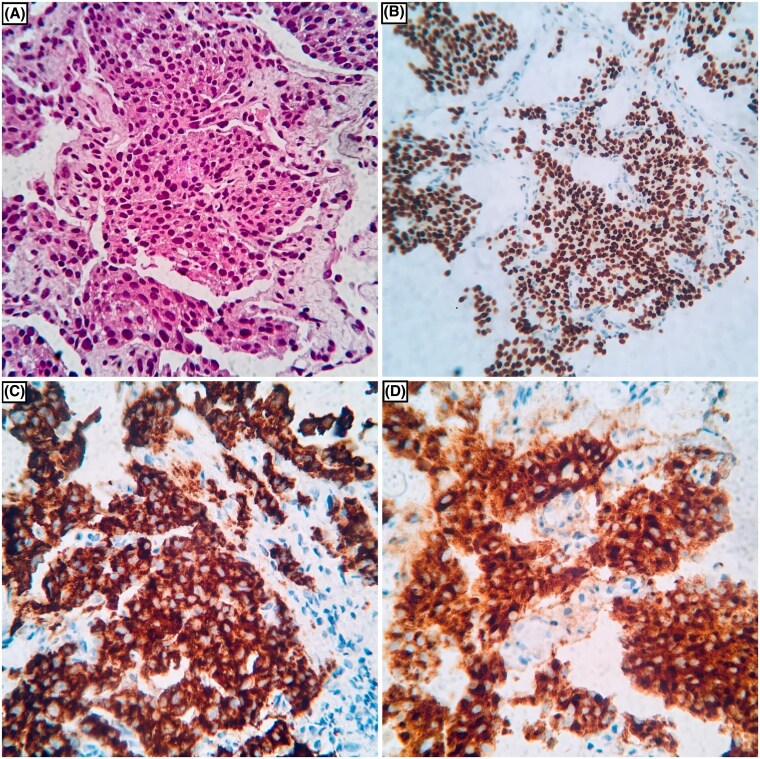 Figure 2
Figure 2| Urine catecholamines | Results (SI units) | Normal range (SI units) |
|---|---|---|
|
| ||
| Metanephrine | 62 μg/24 hours (314.3 nmol/day) | 45-290 μg/24 hours (228.1-1470.3 nmol/day) |
| Normetanephrine | 112 μg/24 hours (611.3 nmol/day) | 75-400 μg/24 hours (409.3-2183.2 nmol/day) |
| Dopamine | 68 μg/24 hours (443.9 nmol/day) | 65-400 μg/24 hours (424.3-2611.2 nmol/day |
|
| ||
| Metanephrine | 191 μg/24 hours (968.4 nmol/day) | 45-290 μg/24 hours (228.1-1470.3 nmol/day) |
| Normetanephrine |
| 75-400 μg/24 hours (409.3-2183.2 nmol/day) |
| Dopamine | 73 μg/24 hours (476.5 nmol/day) | 65-400 μg/24 hours (424.3-2611.2 nmol/day) |
|
| ||
| Metanephrine |
| 45-290 μg/24 hours (228.1-1470.3 nmol/day) |
| Normetanephrine |
| 75-400 μg/24 hours (409.3-2183.2 nmol/day) |
| First author, year of publication [reference] | Demographics: age, gender | Type (PCC/PGL), stage | Catecholamine phenotype | TP53 mutation, classification |
|---|---|---|---|---|
| Hu et al., 2016 [ | 3 yo, female | PCC, local | NA | c.730G>A; p. (Gly244Ser), exon 6, PV |
| Gniado et al., 2020 [ | 39 yo, male | PCC, metastatic | NMN + MN | c.743G>A; p. (Arg248Gln), exon 6, PV |
| Seo et al., 2020 [ | 43 yo, female | PGL, local | NMN + MN | c.566C>T; p. (Ala189Val), exon 6, VUS |
| Choi et al., 2021 [ | 30 yo, female | PGL, metastatic | Functional tumor | c.725G>A; p. (Cys247Tyr), exon 7, PV |
| Choi et al., 2021 [ | 49 yo, female | PCC, metastatic | Functional tumor | c.31G>C p. (Glu11Gln), exon 18, VUS |
| Lima et al., 2023 [ | 32 yo, female | PCC, local | NMN + MN | c.1010G>A; p. (Arg337His), exon 9, PV/LP |
| Stojiljkovic et al., 2024 [ | 19 yo, female | PCC, local | Functional tumor | c.376-2A>G, intronic region (splice acceptor), PV/LP |
| Number [ref.] | Gender (M/F) | Median age (range) | Type (PCC/PGL) (%/%) | Local/metastatic disease (%) | Metastatic sites | Functional status (%) | Other co-mutations (germline/somatic) | |
|---|---|---|---|---|---|---|---|---|
|
|
| 2/6 | 42 | 7/3 | L:5 (50) | Kidneys, LNs | 4/10 | 1 |
|
|
| 1/6 | 37 | 5/2 | L:4 (57.1) | Bones, lungs, LNs | 6/6 | 1 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer-related Molecular Pathways · Cancer, Hypoxia, and Metabolism · Neuroblastoma Research and Treatments
Introduction
Li-Fraumeni syndrome (LFS) is a rare inherited cancer syndrome with an autosomal hereditary pattern. It was first described in 1969, while approximately 20 years later, germline tumor protein p53 (TP53) pathogenic variants were established as the cause of the syndrome [1, 2]. Individuals with LFS bear a markedly increased lifetime risk for neoplasms such as early-onset breast cancer, soft tissue and bone sarcomas, primary central nervous system cancers, acute leukemia and adrenocortical carcinoma, requiring early and strict lifetime surveillance [3]. The TP53 gene, often referred to as the “guardian of the genome,” is dormant under physiological conditions. Cellular stress signals enable the binding of p53 protein to DNA, activating genes responsible for DNA repair, cycle arrest, and apoptosis, aiming to restore normality [4]. This safety brake is abolished by malfunctioning forms of the p53 protein, rendering cells highly vulnerable to DNA damage, ultimately leading to a cascade of tumorigenic events. Most LFS cases are inherited in an autosomal-dominant pattern; however, 7% to 20% of cases are attributed to de novo mutations or mosaicism [5]. Developing a pheochromocytoma/paraganglioma (PCC/PGL) represents a rather unusual presentation in TP53 carriers. We report a female patient with a germline TP53 pathogenic variant who developed metastatic PCC, and we discuss the current literature on this rare situation.
Case Presentation
A 57-year-old woman with LFS was under surveillance at our center. Her past medical history was remarkable for type 2 diabetes mellitus, hypertension, and various malignancies. In 2009, she was diagnosed with papillary thyroid cancer (pT1bN0) and treated with near-total thyroidectomy followed by radioactive iodine therapy. In 2014, a 10-cm asymptomatic adrenal mass was incidentally discovered. Magnetic resonance imaging (MRI) of the abdomen suggested a pheochromocytoma, while suspicious para-aortic lymphadenopathy was noted. The preoperative hormonal profile for catecholamine hypersecretion was negative. The patient underwent surgery, revealing a 9-cm well-circumscribed mass composed of large polygonal cells arranged in nests (Zellballen pattern), a low proliferation index (Ki-67 = 1%-3%), rare mitoses (<2 mitoses/10 high-power fields), absence of necrosis, and low cellularity. Three lymph nodes were negative for metastases. Immunohistochemistry (IHC) was positive for synaptophysin, chromogranin A, S100, and GATA-binding protein 3 (GATA3). The pathology features were consistent with PCC (pT2N0). The Pheochromocytoma of the Adrenal gland Scaled Score (PASS) was 2, and the Grading system for Adrenal Pheochromocytoma and Paraganglioma (GAPP) had a score of 1, both indicating a low probability for metastasis. In 2015, she was diagnosed with high-grade ductal carcinoma in situ (DCIS) of the left breast. She underwent a partial mastectomy followed by radiation therapy and initiated adjuvant endocrine treatment with tamoxifen. A few months later, multiple lesions were identified in the left breast, and a subsequent biopsy showed an invasive breast carcinoma (triple-negative breast cancer). She underwent a left mastectomy and axillary lymph node removal [pT2(m)N1] followed by adjuvant chemotherapy. The presence of multiple malignancies prompted targeted genetic testing (Sanger sequencing) for BRCA1/2 and TP53 gene alterations. Testing revealed a substitution in exon 6 (c.743G>A) of the TP53 gene, resulting in arginine substitution by glutamine at codon 248 (p.Arg248Gln). This missense mutation represents an established pathogenic variant, and the patient was placed on a surveillance program [6]. In 2018, she underwent a prophylactic right mastectomy. Her daughter and 2 sisters tested negative for the TP53 mutation, while her parents have not been assessed due to their advanced age. Her family history includes only one cancer case: her niece, with an unknown genetic background, was diagnosed with dermatofibrosarcoma protuberans.
Diagnostic Assessment
In 2023, her annual whole-body MRI (WBMRI) showed suspicious lung lesions warranting further investigation (Fig. 1A). A subsequent fluorine-18 fluorodeoxyglucose (^18^F-FDG) positron emission tomography/computer tomography (PET/CT) scan revealed multiple bilateral lung nodules (Fig. 1B). Following the imaging results, a CT-guided biopsy demonstrated infiltration by nests of tumor cells with eosinophilic cytoplasm and no mitotic activity. The IHC was positive for GATA3 and the neuroendocrine markers chromogranin A and synaptophysin, and negative for cytokeratin 7 (CK7), anti-cytokeratin 5.2 (CAM 5.2), cytokeratin AE1/3, cytokeratin 5/6 (CK5/6), thyroid transcription factor 1 (TTF-1), tumor protein 63 (p63), and estrogen receptor (ER). These findings were compatible with metastatic PCC (Fig. 2). Given the neuroendocrine nature of the neoplasm, a gallium-68 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA)–octreotate (^68^Ga-DOTATATE) PET/CT scan was performed. It confirmed the presence of somatostatin receptor-expressing tumor tissue in the lungs and in the first lumbar vertebrae (L1) (Fig. 1C and 1D). Clinically, the patient did not report any signs or symptoms due to excessive catecholamine secretion. A 24-hour urine collection for fractionated metanephrine, normetanephrine and 3-methoxytyramine was ordered, and the results were within normal limits (Table 1). Moreover, new germline testing was performed using a next-generation sequencing (NGS) assay (NextSeq 2000, Illumina, Inc.), interrogating a broad spectrum of genes related to PCCs/PGLs (EGLN1, FH, MAX, MEN1, NF1, RET, SDHA, SDHA2, SDHB, SDHC, SDHD, TMEM127, VHL). Testing was negative for any germline mutations other than TP53. The tissue sample was tested for additional somatic mutations (NextSeq 2000, Illumina, Inc.) and copy number variations (MLPA®, MRC Holland) with negative results.
Imaging techniques were used during the case investigation. A. MRI of the abdomen (T2-weighted axial view) capturing the lower lung fields showed suspicious lesions (white circles). B. Fluorine-18 fluorodeoxyglucose PET/CT scan (axial view) depicted bilateral lung nodules (white circles). C. 68Ga-DOTATATE PET/CT scan (axial view) revealed somatostatin receptor-expressing lung lesions (white circles). D. SSTR-expressing tissue was also detected in the body of L1 by 68Ga-DOTATATE PET/CT scanning (axial view, white circle).
Histopathology images of the lung lesion. (A) Tumor cells arranged in nests (Zellballen pattern), hematoxylin and eosin-stained section at 200×. (B) Tumor cells showed strong GATA3 positivity. (C) Synaptophysin and (D) Chromogranin A staining are diffusely positive.
Treatment
Given the absence of symptoms and signs and no evidence of catecholamine overproduction, the patient continued with follow-up and strict surveillance. A few months later, a new 24-hour urine test showed an increase in metanephrine levels, leading to the decision to start the patient on subcutaneous lanreotide autogel, 120 mg monthly. A WBMRI did not demonstrate any radiological disease progression. Subsequent repeat testing indicated a further rise in 24-hour urinary metanephrine levels, accompanied by blood pressure dysregulation. Antihypertensive therapy was modified to include doxazosin, 2 mg daily.
Outcome and Follow-Up
The patient continues to be closely monitored for signs and symptoms of catecholamine excess. At present, she is asymptomatic, and her blood pressure is well-controlled. She undergoes regular WBMRI scanning; in due course, ^68^Ga-DOTATATE PET/CT will be repeated. In case of disease progression, further treatment options include radioisotope therapy with peptide receptor radionuclide therapy (PRRT) or iodine-131 meta-iodobenzylguanidine (^131^I-MIBG), systemic chemotherapy (cyclophosphamide-vincristine-dacarbazine, or temozolomide monotherapy), and molecular targeted therapy (cabozantinib, sunitinib).
Discussion
PCCs/PGLs are rare neuroendocrine tumors arising from the adrenal medulla or extra-adrenal paraganglia. They are increasingly being discovered incidentally and predominantly produce catecholamines responsible for their symptomatology [7]. According to the latest World Health Organization (WHO) classification, virtually all PCCs/PGLs have metastatic potential, and the terms benign and malignant are no longer advised [8].
PCCs/PGSs have one of the highest genetic predispositions among all tumors, and international guidelines strongly endorse genetic testing. Approximately 30% to 40% of cases are associated with inherited syndromes harboring a germline mutation in genes related to PCCs/PGLs, while an additional 35% to 40% of tumors bear a somatic driver mutation [9]. Intriguingly, in 10% to 13% of seemingly sporadic cases, a germline pathogenic mutation in a culprit genetic locus is detected [10, 11]. Recent advances in molecular biology have enabled the in-depth characterization of the molecular background of these neoplasms. PCCs/PGLs can be assigned to 1 of 3 clusters based on the molecular pathway of tumorigenesis. Cluster 1, or the pseudo-hypoxic cluster, comprises genes that activate pathways resembling hypoxia signaling: these genes include SDHA-D, FH, MDH2, SLC25A11, and VHL, and are predominantly correlated with germline mutations. Cluster 2 includes genes implicated in tyrosine kinase signaling, such as RET, HRAS, NF1, TMEM127, MAX, and FGFR1. Mutations for some genes could be either germline or somatic, while for others, they are exclusively somatic. Cluster 3 is related to β-catenin/Wnt-signaling activation and comprises the MAML3 fusion gene and CSDE1; mutations in this group are solely somatic [12].
The genetic landscape of PCCs/PGLs has largely been deciphered, but associations with TP53 mutations have not been frequently observed. LFS appears to be strongly related to numerous types of cancer; however, the connection with PCCs/PGLs is infrequent and is only noted in sporadic case reports (Table 2). In 2016, Hu et al reported the first Chinese family with LFS, where one of the affected members was diagnosed with PCC at the age of 3 years [13]. A case of metastatic PCC in the context of LFS was reported by Gniado et al in 2021; however, the authors noted that the patient also carried another germline mutation in the SDHB gene, which is well known to be associated with PCC [14]. Seo et al reported a single TP53 germline mutation in 36 patients with PCCs/PGLs [15]. Two cases of germline TP53 mutations were reported in a North Korean series of 15 cases [16]. In a series of 101 PCCs/PGLs reported by Lima et al, there was one case of a germline TP53 carrier, but the authors stated that the patient also had a germline pathogenic mutation in the TMEM127 gene [17]. A case from Serbia was published regarding a carrier of a germline TP53 pathogenic variant with a history of PCC [18].
As noted above, extensive molecular profiling has greatly illuminated the genomic blueprint of PCCs/PGLs. Although the loss of the short arm of chromosome 17, which contains the genetic locus of TP53, is considered a frequent genomic event in PCCs/PGLs, more specific TP53 genomic alterations have also been identified [10, 19]. The French COMETE cohort was one of the first initiatives aiming to shed light on the genomic landscape of PCCs/PGLs. A multi-omics analysis of 202 samples revealed somatic mutations in the TP53 gene in only 2 cases [20]. Another initiative was launched by the Cancer Genome Atlas (TCGA) research network by conducting a comprehensive molecular characterization of 173 PCCs/PGLs. No germline TP53 germline pathogenic variants were detected, while somatic alterations were found in just one case [10]. A large cohort conducted in the United Kingdom that included 141 individuals with PCCs/PGLs aimed to investigate the clinical utility of somatic sequencing: 45 patients harbored germline mutations, but none involving the TP53 gene, and while 37 had pathogenetic somatic mutations, only one referred to TP53 [21]. Other smaller studies have confirmed the rarity of germline and somatic TP53 mutations [16, 22-24].
Searching the literature, we detected 7 germline TP53 variants (0.74%) in 942 PCCs/PGLs with germline investigation [10, 13-18, 20-24], while of 788 cases with somatic testing, 10 bore somatic variants (1.27%) [10, 16, 20-24]. Table 3 summarizes the characteristics of PCCs/PGLs with TP53 alterations. Most carriers with germline mutations are female and tend to develop PCCs (71.4%), which are functional tumors (100%) with mixed catecholamine secretion profiles. When metastatic, the tumors tended to metastasize primarily to bones and lungs. In 2 cases, germline co-mutations with SDHB and TMEM127 genes were detected. Both genes are related to the development of PCCs/PGLs, and the contribution of the TP53 mutation to tumorigenesis in this context is unclear.
In our case, the knockout of the wild-type TP53 allele, leading to loss of heterozygosity, could represent a potential tumorigenesis mechanism. However, molecular investigation of the tumor did not reveal a deleterious mutation or loss of the genetic locus of the wild-type TP53 gene; thus, epigenetic silencing of the wild-type TP53 allele could be speculated as a possible mechanism. Geli et al showed that PCCs/PGLs exhibit an extensive epigenomic dysregulation, leading to epigenetic silencing of tumor suppressor genes and correlating with aggressive disease [25]. Furthermore, mutations in genes related to epigenetic regulation, such as ATRX, SETD2, and KMT2D, have been described in PCCs/PGLs [12]. It has also been demonstrated that PCCs/PGLs with gene mutations included in Cluster 1 exhibit a profound hypermethylation phenotype [26]. Another theory supports the concept that deleterious germline TP53 mutations exert a predominantly dominant-negative effect on the wild-type TP53 allele, abrogating normal protein function [27].
Our patient was exposed to radiation following the diagnosis of DCIS, and a few months later she developed invasive carcinoma. Although the evidence regarding the impact of radiation on TP53 carriers is scarce, radiotherapy appears to further increase the risk of secondary malignancies, particularly for soft tissue sarcomas [28]. PRRT is an established treatment for PCCs/PGLs. Typically, the recommended schedule for PRRT with lutetium-177 (^177^Lu)-DOTATATE consists of 4 fixed cycles of 7.4 GBq (200 mCi) infusions every 6 to 12 weeks. Nevertheless, the effects of the emitted radiation from this type of therapy in LFS individuals remain unknown, and its possible future use in this patient would need to be more carefully considered than in patients with other types of mutation.
Learning Points
LFS is associated with a broad spectrum of malignancies. PCCs/PGLs represent infrequent manifestations of the syndrome (≈1%).Carriers of deleterious germline TP53 variants should be closely monitored from a young age. Screening recommendations propose comprehensive physical examination, ultrasound of the abdomen and pelvis every 3 to 4 months, and annual WB-MRI for carriers starting at birth.Based on the existing literature, we recommend periodic blood pressure monitoring and consideration of assessments of metanephrines secretion wherever there is clinical suspicion. Where positive, one could proceed to careful evaluation of WBMRI for adrenal lesions and ^68^Ga-DOTATATE PET/CT scanning may be useful.
Contributors
All authors made individual contributions to authorship. S.L., C.M., and A.G. were involved in the diagnosis and management of this patient; E.I. was involved in histopathology pictures presentation; and K.P. and A.G. were involved in manuscript editing and correction. All authors reviewed and approved the final draft.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Li FP, Fraumeni JF Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71(4):747‐752.5360287 10.7326/0003-4819-71-4-747 · doi ↗ · pubmed ↗
- 2Malkin D, Li FP, Strong LC, et al Germ line p 53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250(4985):1233‐1238.1978757 10.1126/science.1978757 · doi ↗ · pubmed ↗
- 3Schneider K, Zelley K, Nichols KE, Schwartz Levine A, Garber J. Gene Reviews: Li-Fraumeni Syndrome. University of Washington; 1993.20301488 · pubmed ↗
- 4Aubrey BJ, Strasser A, Kelly GL. Tumor-suppressor functions of the TP 53 pathway. Cold Spring Harb Perspect Med. 2016;6(5):a 026062.27141080 10.1101/cshperspect.a 026062 PMC 4852799 · doi ↗ · pubmed ↗
- 5Renaux-Petel M, Charbonnier F, Théry JC, et al Contribution of de novo and mosaic TP 53 mutations to Li-Fraumeni syndrome. J Med Genet. 2018;55(3):173‐180.29070607 10.1136/jmedgenet-2017-104976 · doi ↗ · pubmed ↗
- 6Clin Var . National Center for Biotechnology Information. Updated April 20, 2025. Accessed April 21, 2025. https://www.ncbi.nlm.nih.gov/clinvar/variation/12356/
- 7Aggarwal S, Prete A, Chortis V, et al Pheochromocytomas most commonly present as adrenal incidentalomas: a large tertiary center experience. J Clin Endocrinol Metab. 2023;109(1):e 389‐e 396.37417693 10.1210/clinem/dgad 401PMC 10735286 · doi ↗ · pubmed ↗
- 8Mete O, Asa SL, Gill AJ, Kimura N, de Krijger RR, Tischler A. Overview of the 2022 WHO classification of paragangliomas and pheochromocytomas. Endocr Pathol. 2022;33(1):90‐114.35285002 10.1007/s 12022-022-09704-6 · doi ↗ · pubmed ↗