Metagenomic Identification of a Novel Zoonotic Gemykibivirus in a Diarrheic Pig in China
Wenqiang Wang, Lin Yuan, Miaojie Zhang, Qilin Zhao, Wenqiang Pang, Dehu Sun, Xiaoye Jia, Feifei Tan, Tingting Niu, Kegong Tian, Xiangdong Li

TL;DR
A new gemykibivirus was found in a sick pig in China, suggesting it could spread between animals and humans.
Contribution
The discovery of a novel zoonotic gemykibivirus in pigs expands understanding of cross-species transmission and evolution.
Findings
A novel gemykibivirus (pGkV) was identified in a diarrheic pig in China.
pGkV is closely related to human and avian gemykibiviruses, indicating potential zoonotic transmission.
Recombination signals suggest recombination contributes to the emergence of new gemykibivirus strains.
Abstract
Gemykibiviruses are circular, replication-associated protein (REP) encoding single-stranded DNA (ssDNA) viruses classified within the genus Gemykibivirus of the family Genomoviridae. In recent years, several gemykibiviruses have been detected in humans presenting with clinical symptoms, such as encephalitis, respiratory illness, sepsis, pericarditis, diarrhea, and multiple sclerosis. However, the presence of gemykibiviruses in other animal hosts, as well as the evolution of the Gemykibivirus genus, remains poorly understood. In this study, we identified a novel gemykibivirus from a diarrheic pig in China using metagenomic sequencing, which we designated as pGkV. The pGkV genome comprises 2200 nucleotides and encodes two key proteins as follows: the capsid-associated protein (CAP) and the REP. Phylogenetic analyses revealed that pGkV clusters within the zoonotic gemykibivirus (ZooGkV)…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
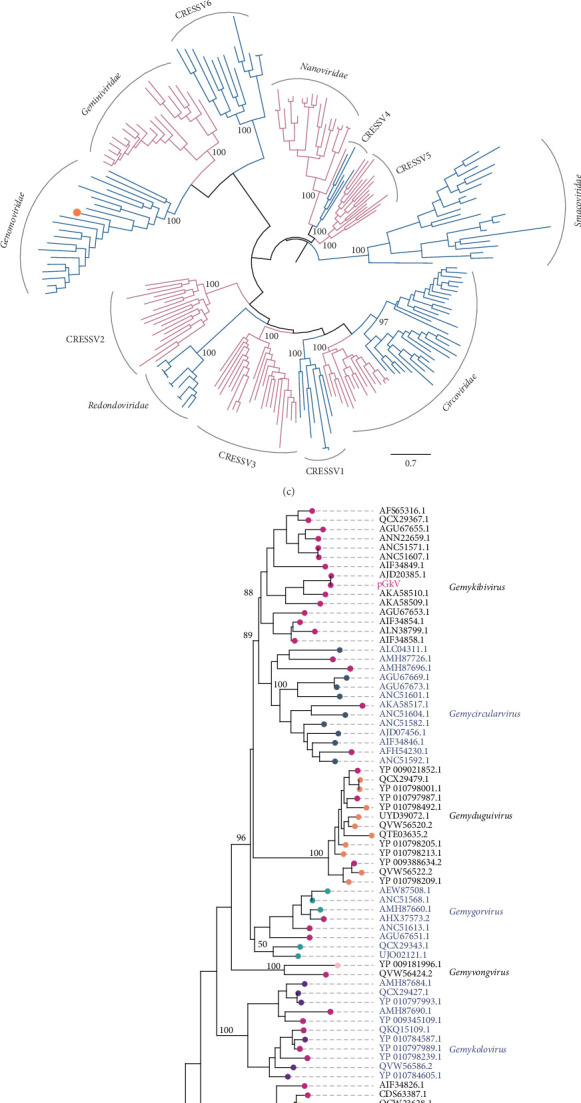 Figure 1
Figure 1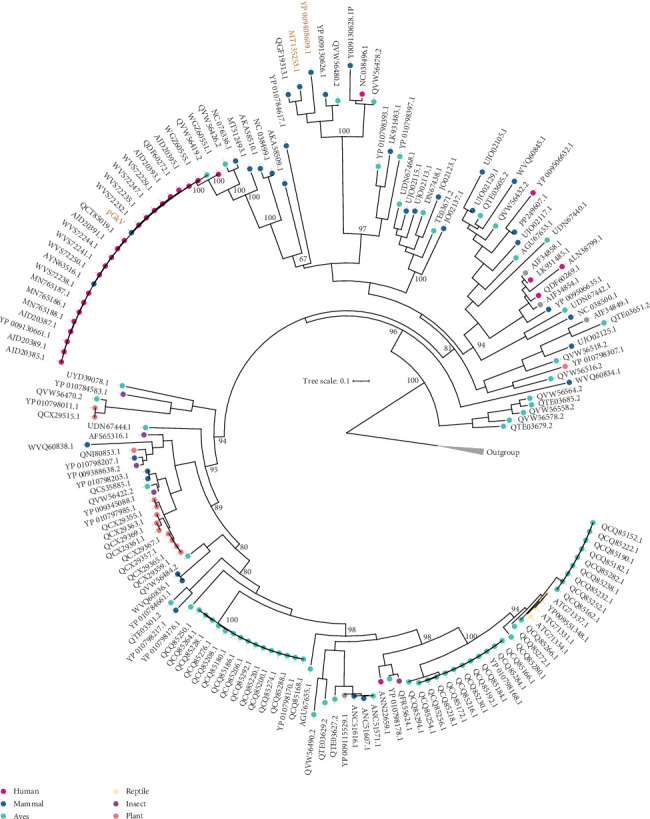 Figure 2
Figure 2 Figure 3
Figure 3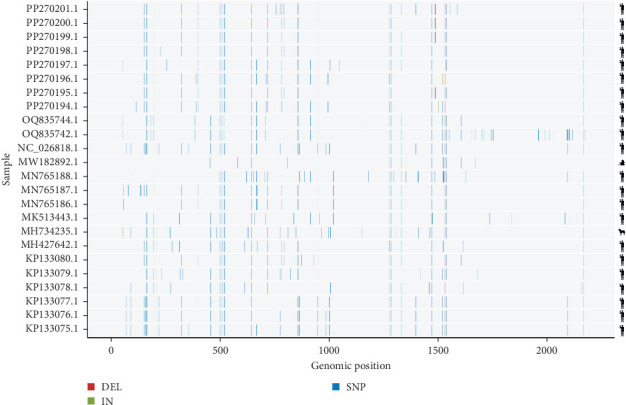 Figure 4
Figure 4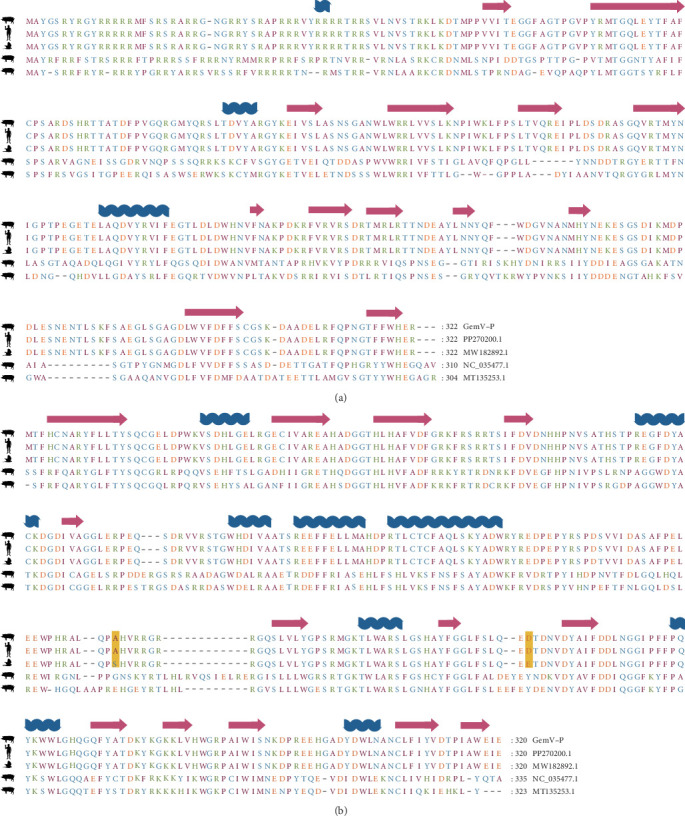 Figure 5
Figure 5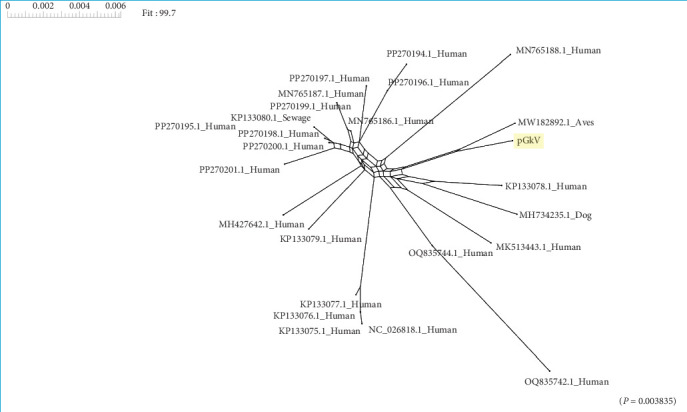 Figure 6
Figure 6- —National Key Research and Development Program of China
- —Jiangsu Innovative and Entrepreneurial Talent Team Project
- —111 Project D18007
- —Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Virus Research Studies · Animal Virus Infections Studies · Viral gastroenteritis research and epidemiology
1. Introduction
Metagenomic next-generation sequencing (mNGS) has substantially enhanced the efficiency of novel pathogen discovery due to its nontargeted, high-throughput nucleic acid detection capabilities, particularly in cases of complex infections and outbreaks caused by unknown pathogens [1]. This was exemplified by its pivotal role in the rapid identification of SARS-CoV-2 during the COVID-19 pandemic, with the virus being characterized within 40 h [2]. By enabling unbiased detection of both known and previously unrecognized viruses through comprehensive nucleic acid sequencing, mNGS has greatly expanded our understanding of viral diversity [3, 4].
The family Genomoviridae, established by the International Committee on Taxonomy of Viruses (ICTV) in 2016, constitutes a distinct lineage of circular Rep-encoding single-stranded DNA (CRESS-DNA) viruses [5]. These viruses are characterized by small, monopartite, nonenveloped genomes encapsidated within icosahedral particles measuring approximately 20–22 nm in diameter. Genomic analyses have revealed that members of this family possess compact circular ssDNA genomes ranging from 1.8 to 2.4 kilobases (kb) in length [5]. Currently, Genomoviridae comprises 10 genera, including Gemykibivirus, Gemycircularvirus, Gemyduguivirus, Gemygorvirus, Gemyvongvirus, Gemykolovirus, Gemykrogvirus, Gemytripvirus, Gemykroznavirus, and Gemytondvirus [5, 6]. These viruses typically encode two major open reading frames (ORFs): one for a replication-associated protein (REP), which contains conserved endonuclease and helicase domains, and another for a capsid protein (CAP), often expressed via spliced transcripts. Phylogenetically, the Genomoviridae family forms a sister clade to the Geminiviridae, with species demarcation defined by less than 78% whole-genome sequence identity [6].
Viruses of the Gemykibivirus genus have been predominantly detected in various human clinical cases, across a wide range of sample types, including those associated with encephalitis, respiratory diseases, sepsis, pericarditis, diarrhea, and multiple sclerosis [7–9]. However, a direct causal relationship between gemykibivirus infection and these clinical manifestations has not been firmly established. The occurrence of gemykibiviruses in nonhuman hosts and the mechanisms underlying their cross-species transmission remain poorly understood. In this study, we identified a novel gemykibivirus from a diarrheic pig in China using mNGS technology. We further conducted a comprehensive evolutionary analysis of viruses within the Gemykibivirus genus. Our findings provide new evidence supporting the potential zoonotic capacity of this newly identified porcine gemykibivirus (pGKV), highlighting the importance of continued surveillance, and characterization of gemykibiviruses in both human and animal hosts.
2. Materials and Methods
2.1. Sample Collection, DNA Extraction, and Sequencing
Tissue sample was homogenized in sterile PBS (90 Hz, 90s) and centrifuged at 3000 rpm for 5 min. A 0.2 mL aliquot of the supernatant was collected, with porcine kidney cells (PK-15) serving as the negative control. Total DNA and RNA were coextracted using the MagMAX CORE Nucleic Acid Extraction Kit via magnetic bead-based purification. Nucleic acid concentrations were quantified using the Equalbit 1×dsDNA HS Assay Kit and Equalbit RNA HS Assay Kit. Library construction was performed according to the protocol provided by the manufacturer of the MGIEasy Fast Enzymatic Fragmentation Library Prep Kit v2.0. DNA nanoballs (DNBs) were prepared using the MGISEQ-2000RS High-Throughput Rapid Sequencing Kit. Sequencing was conducted on the MGISEQ-2000 platform with a single-end 100 bp (SE100) read length.
2.2. Data Processing
Sequencing reads were first adaptor- and quality-trimmed using the Fastp (v 0.23.2) program with default parameters [10]. After quality control, the clean data were further aligned to the pig genome (GCF_000003025.6) to remove the host genome using bowtie2 (v2.3.5.1) program [11]. The remaining reads were assembled de novo using SPAdes (v3.15.5) program with default parameter settings [12]. Assembled contigs were compared against the NCBI core nucleotide database (core nt) using online BLASTn. A CRESSV-like virus with genome length 2200 nucleotides were identified.
2.3. Phylogenetic Analyses
In order to further elucidate the new identified CRESSV belongs to which CRESSV family, we integrated the REP protein of the new virus with the REP sequences from various families, including Circoviridae, Smacoviridae, Nanoviridae, Geminiviridae, Genomoviridae, Redondoviridae, and CRESSV1~6. The virus sequences were aligned using the L-INS-i algorithm implemented in the program MAFFT (v7.518) [13]. Maximum-likelihood phylogenies were inferred from the amino acid alignments using IQ-TREE (v 2.2.0) under the best-fit substitution model, automatically selected via the -m MFP option. Branch support was assessed with 1000 replicates of the SH-like approximate likelihood ratio test (SH-aLRT) [14]. Upon confirming the classification of the novel virus within the Genomoviridae family, we proceeded to further characterize and validate its genus-level taxonomic assignment. We selected represent sequences from all 10 genus of Genomoviridae. MAFFT (v7.518) and IQ-TREE (v 2.2.0) were used to align REP protein sequences and constructed phylogenetic relationships, respectively. In order to investigate the evolution of viruses in genus Gemykibivirus and identify the phylogenetic position of the novel virus among the genus, we download all Gemykibivirus sequences from genbank in NCBI and further validated the taxonomic assignment of these Gemykibiviruses by phylogenetic analyses. The REP protein sequences of all characterized Gemykibiviruses, including the novel isolate, were aligned using MAFFT and subsequently employed to construct a maximum-likelihood phylogenetic tree with IQ-TREE.
2.4. Mutation and Haplotype Analyses
To evaluate genomic similarity among zoonotic gemykibiviruses (ZooGkVs), we performed pairwise alignments between the novel virus, which served as the reference genome, and each of the other known Zoo-GkVs using MAFFT [13]. Genetic variations, including single-nucleotide polymorphisms (SNPs), insertions, and deletions, were identified and compiled into a variant call format (VCF) file using a custom Python script. Subsequently, a genome-wide mutation map was visualized using an R-based package. Haplotype network analyses were also employed in this study. The genome sequences of all Zoo-GkVs were aligned using MAFFT, and the resulting multiple sequence alignment file was subsequently analyzed in DNAsp to generate a haplotype data file [15]. Haplotype network analysis was then performed in POPART utilizing the median-joining algorithm [16].
2.5. Recombination Analyses
To elucidate the role of recombination in the Zoo-GkVs, we conducted comprehensive recombination analyses across all Zoo-GkVs. The multiple sequence alignments originally constructed for haplotype characterization were repurposed for recombination detection analyses in the current investigation. To detect potential recombination events, we employed SplitsTree software to construct phylogenetic networks for visualizing recombination signals [17]. Additionally, we performed Pairwise Homoplasy Index (PHI) tests to statistically assess recombination events across the genome, with a significance threshold of p < 0.05 for identifying evolutionarily significant recombination patterns.
3. Results
3.1. Discovery and Genome Annotation of a New Virus in Swine
mNGS has become an increasingly important tool in pathogen identification by enabling the comprehensive analysis of microbial and host genetic material within a given sample. In this study, mNGS was employed to identify pathogens in a diarrheic pig, leading to the complete sequencing of a new circular single-stranded DNA virus genome. The viral genome is 2200 nucleotides in length and features a nonanucleotide motif [TAAAATTTA], rich in adenine (A) and thymine (T), at the apex of the stem-loop structure (Figure 1). Gene annotation revealed that the circular virus harbors two genes arranged in opposite orientations; one encoding the CAP protein on the positive strand and the other encoding the REP protein on the negative strand, with an intron located in the rep gene (Figure 1A). The genomic features are consistent with those of CRESS-DNA viruses, supporting the classification of this virus within the CRESS-DNA group.
3.2. Phylogenetic Analyses of Gemykibiviruses
Given that porcine circoviruses, also members of the CRESS DNA virus group, have caused substantial economic losses in the pig industry, determining the phylogenetic position of the newly identified CRESS DNA virus is of particular relevance. To this end, we selected 216 REP protein sequences from 12 major CRESS DNA virus families, including the families Circoviridae, Redondoviridae, Genomoviridae, Geminiviridae, Nanoviridae, Smacoviridae, and CRESSV1~6, and integrated them with the Rep sequence of the CRESS DNA virus identified in this study to construct a phylogenetic tree. Phylogenetic analysis revealed that the newly identified CRESS DNA virus and the members of the Genomoviridae family form a stable monophyletic group, strongly supported by a bootstrap value of 100 (Figure 1C). Upon confirming the virus's affiliation with Genomoviridae family, we selected 84 representative REP protein sequences from this family, covering all genera within the family, and combined them with the new virus identified in this study to construct the phylogenetic tree. Results demonstrated that the new virus clusters within the Gemykibivirus genus with a bootstrap value of 88 (Figure 1D). Based on the phylogenetic findings, we have designated the newly identified CRESS DNA virus as pGkV.
To advance our understanding of the diversity within the Gemykibivirus genus and to clarify the taxonomic position of pGkV, we constructed a phylogenetic tree based on REP protein sequences from all gemykibiviruses reported to date. The analysis revealed that the Gemykibivirus genus exhibits a broad host spectrum, encompassing humans, other mammals, birds, reptiles, insects, and plants (Figure 2). Gemykibiviruses from different host groups consistently formed distinct, stable monophyletic clusters within the phylogenetic tree. This widespread distribution of monophyletic groups underscores not only the significant diversity within the Gemykibivirus genus but also its strong potential for cross-host transmission (Figure 2). Notably, pGkV, identified in this study, is not closely related to the two previously reported pig gemykibiviruses. Instead, it clustered with a group of human gemykibiviruses with strong phylogenetic support (bootstrap value = 100), and exhibited short branch lengths within the clade, indicating a close genetic relationship among these gemykibiviruses (Figure 2). We designated this group as the “Zoo-GkV”. These findings suggest that pGkV likely represents a novel member of this emerging zoonotic lineage.
3.3. Evolutionary Insights and Cross-Host Affinities of pGkV
Due to the high sequence similarity of REP proteins among Zoo-GkV members, the precise phylogenetic placement of pGkV within this clade could not be fully resolved. To further elucidate the evolutionary relationship between pGkV and other Zoo-GkV members, we constructed a haplotype network based on SNPs derived from whole-genome alignments. The analysis revealed that pGkV is most closely related to a bird-associated gemykibivirus (Figure 3). To gain a more detailed view of the genomic variation, we performed pairwise genome alignments using pGkV as the reference, providing a comprehensive overview of sequence divergence between pGkV and other Zoo-GkV strains. The mutation landscape revealed that the first half of the pGkV genome exhibits the highest similarity to a bird-derived gemykibivirus, differing by only five SNPs, while the latter half shows greater similarity to several human-associated gemykibiviruses (Figure 4). We then compared the CAP and REP proteins of pGkV with four representative gemykibiviruses and found that neither the CAP nor the REP protein of pGkV showed high similarity to the previously reported swine viruses, consistent with the earlier phylogenetic results (Figure 5). Although haplotype network displays that pGkV is most closely related to the bird-associated gemykibivirus, protein alignments reveal that the REP and CAP proteins are completely identical to the human gemykibivirus, further indicating the zoonotic ability of pGkV (Figure 5). The CAP protein of the bird-associated gemykibivirus is identical to that of pGkV, whereas its REP protein differs by two amino acid residues (Figure 5).
3.4. Recombination Contributes to the Emergence of New Gemykibiviruses
Given that the haplotype network indicates that pGkV is most closely related to the bird-associated gemykibivirus, while protein alignments reveal that proteins of pGkV are absolutely identical to the human gemykibivirus, we infer that the Zoo-GkVs may have experienced recombination. To validate the hypothesis, we performed recombination analyses across these Zoo-GkVs. Recombination analysis revealed a distinct network topology among Zoo-GkVs, indicating the occurrence of recombination events (Figure 6). This finding was further substantiated by the PHI test, which confirmed the statistical significance of these recombination events (p=0.0038).
4. Discussion
In this study, we employed mNGS to identify a gemykibivirus (pGkV) from a diarrheic pig in China. The complete genome sequence of pGkV has been deposited in the NCBI GenBank database under the Accession Number PV329706.1. The identified pGkV is not closely related to the two previously reported pGkVs, but instead forms a stable monophyletic group with some human gemykibiviruses with a bootstrap value of 100, suggesting that pGkV is a new gemykibivirus and not derived from the previously reported pGkVs (Figure 2). The pGkV related cluster was named Zoo-GkV, which also contains a dog associated gemykibivirus and a bird associated gemykibivirus. Zoo-GkV contains most of the human-associated gemykibiviruses reported to date, which were mainly identified in patients with some symptoms, including encephalitis, diarrhea, febrile, and acute respiratory distress syndrome [18–20]. Regrettably, although many gemykibiviruses were identified in patients with obvious symptoms, direct evidence supporting members of the genus Gemykibivirus as causative agents of disease is still lacking. In this study, the pGkV-positive sample also contained other viruses, including African swine fever virus, adenovirus, and coronavirus, indicating a coinfection scenario. Hence, whether the diarrhea observed in the pig was caused by pGkV remains unclear.
Although the haplotype network displayed that pGkV is most closely related to the bird associated gemykibivirus (MW182892.1), the two encoded proteins, REP and CAP, are completely identical to those of a human gemykibivirus (PP270195.1), indicating that pGkV may possess the ability to infect humans without requiring any genetic variation (Figure 5). This raises the possibility of cross-species transmission, although the exact routes and mechanisms remain to be determined. The identification of Zoo-GkV in patients from Nepal, China, France, and Brazil suggests that gemykibiviruses may be globally disseminated [18, 20–22]. Zoo-GkV was initially detected in 2017 in China, where it was identified in a throat swab specimen collected from a woman suffering from unexplained acute respiratory distress syndrome [20]. Together with the identification of pGkV in China, these data indicate that the Zoo-GkVs have been circulating in China for nearly a decade, and even exhibit different host tropisms. In addition, we detected pGkV-related reads in another clinical sample, although a complete genome could not be assembled, suggesting that pGkV may not be an isolated case. With the help of metagenomic NGS, an increasing number of gemykibiviruses are expected to be discovered in the future.
Genome and recombination analyses reveal recombination events in Zoo-GkV, which provide genetic material for the evolution of gemykibiviruses and may act as an important driving force for the emergence of new gemykibivirus strains. Given the fact that coinfection with multiple viruses is the prerequisite for recombination, the existence of recombination events in Zoo-GkV indicates occurrence of gemykibivirus coinfection. The phylogenetic tree of gemykibiviruses constructed in this study is the most detailed representation of the Gemykibivirus genus to date, and this genus comprises viruses that have been detected in various parts of the world, associated with various organisms, including mammals, birds, reptiles, insects, and plants. Notably, the host range of the Zoo-GkV lineage includes both mammals and aves. Recombination analyses were employed only across the Zoo-GkVs in this study, and more recombination events may exist in the broader Gemykibivirus genus.
One limitation of this study is that the pGkV was not successfully isolated, despite attempts using standard protocols for circovirus cultivation, thereby limiting our knowledge of the pathogenic role of pGkV. Not only has pGKV not been successfully isolated, but no members of the Gemykibivirus genus have been isolated and utilized in animal experimentation to date. Hence, there is an urgent need to isolate gemykibivirus and elucidate transmission routes, host interactions and pathogenesis. Although a clear lack of direct evidence supporting the genus Gemykibivirus as a potential candidate for disease causation exists, many gemykibiviruses were identified in patients and animals with diseases, indicating their potential pathogenicity [18–20, 23]. Nevertheless, this study identified a new gemykibivirus and assessed its potential zoonotic risk, providing novel insight into the evolution of Gemykibivirus genus.
5. Conclusion
In this study, we identified and characterized a novel gemykibivirus, designated pGkV, from a diarrheic pig in China. Genomic and phylogenetic analyses demonstrated that pGkV belongs to the Zoo-GkV clade and shares high similarity with both avian and human strains, particularly in its CAP and REP protein sequences, highlighting its potential zoonotic nature. Moreover, the detection of recombination signals among Zoo-GkVes points to recombination as a potential evolutionary mechanism driving the emergence of novel strains. Together, our results expand the known host range of gemykibiviruses and provide important insights into their evolution and cross-species transmission dynamics, underscoring the need for enhanced surveillance of these viruses in both human and animal populations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chiu C. Y. Miller S. A. Clinical Metagenomics Nature Reviews Genetics 201920634135510.1038/s 41576-019-0113-72-s 2.0-85063607904 PMC 685879630918369 · doi ↗ · pubmed ↗
- 2Wu F. Zhao S. Yu B. A New Coronavirus Associated With Human Respiratory Disease in China Nature 2020579779826526910.1038/s 41586-020-2008-332015508 PMC 7094943 · doi ↗ · pubmed ↗
- 3Chen Y. M. Hu S. J. Lin X. D. Host Traits Shape Virome Composition and Virus Transmission in Wild Small Mammals Cell 20231862146624675.e 461210.1016/j.cell.2023.08.02937734372 · doi ↗ · pubmed ↗
- 4Hou X. He Y. Fang P. Using Artificial Intelligence to Document the Hidden RNA Virosphere Cell 20241872469296942.e 691610.1016/j.cell.2024.09.02739389057 · doi ↗ · pubmed ↗
- 5Krupovic M. Ghabrial S. A. Jiang D. Varsani A. Genomoviridae: A New Family of Widespread Single-Stranded DNA Viruses Archives of Virology 201616192633264310.1007/s 00705-016-2943-32-s 2.0-8497631602327343045 · doi ↗ · pubmed ↗
- 6Varsani A. Krupovic M. Sequence-Based Taxonomic Framework for the Classification of Uncultured Single-Stranded DNA Viruses of the Family Genomoviridae Virus Evolution 20173110.1093/ve/vew 037vew 037PMC 539992728458911 · doi ↗ · pubmed ↗
- 7Halary S. Duraisamy R. Fancello L. Novel Single-Stranded DNA Circular Viruses in Pericardial Fluid of Patient With Recurrent Pericarditis Emerging Infectious Diseases 201622101839184110.3201/eid 2210.1600522-s 2.0-8498853015327648613 PMC 5038422 · doi ↗ · pubmed ↗
- 8Macera L. Spezia P. G. Medici C. Low Prevalence of Gemycircularvirus DNA in Immunocompetent and Immunocompromised Subjects The New Microbiologica 201942211812031034081 · pubmed ↗