Proto‐Oncogene HRAS Transcript Level and Overall Survival in Stages II and III Colorectal Cancer
Donghyun Kim, Saima Sharif, Juan Antonio Raygoza Garay, Avanish S. Bhakta, Patrick M. Boland, Michael J. Cavnar, Michelle L. Churchman, Hassan Hatoum, Lyen C. Huang, Joseph Kim, Richard Kim, Robert W. Lentz, Sarbajit Mukherjee, Mary T. O'Donnell, Benjamin Quartey

TL;DR
High levels of HRAS gene activity in colorectal cancer are linked to better survival, especially in patients with low KRAS activity and no KRAS mutations.
Contribution
This study reveals that high HRAS transcript levels are associated with improved survival in stages II and III colorectal cancer.
Findings
High HRAS transcript levels correlate with superior overall survival in stages II and III CRC.
The survival benefit of high HRAS is most notable in right-sided CRC with low KRAS transcript levels and no KRAS mutations.
Abstract
Mutational landscape is prognostic in colorectal cancer (CRC). Rat sarcoma (RAS) oncogenes, such as KRAS and NRAS, with driver mutations, portend poor survival outcomes, whereas pathologic mutations in HRAS are extremely rare, and their prognostic value remains uncertain. This retrospective study analyzed the Oncology Research Information Exchange Network (ORIEN) alliance tumor RNA‐Seq data in Stages II and III CRC to investigate the association between RAS gene expression and survival outcomes. High transcript levels of HRAS were associated with superior overall survival (OS). The high HRAS‐associated OS benefit was most pronounced in patients with right‐sided primary expressing low KRAS transcript levels in the absence of pathologic KRAS mutations. Contrary to the notion that RAS family genes are proto‐oncogenic, this study demonstrates that high HRAS transcript levels are…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
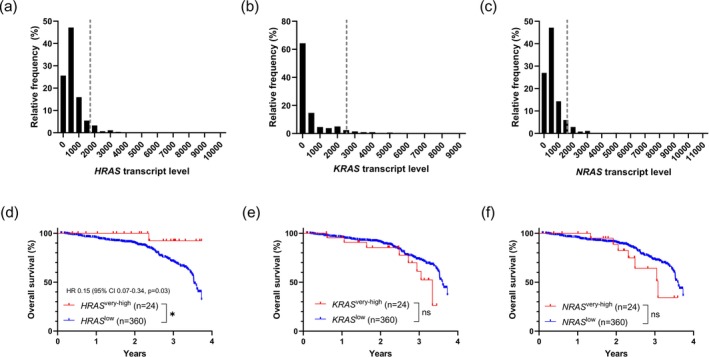 FIGURE 1
FIGURE 1| Variable | HR | 95% CI |
|
|---|---|---|---|
| Gender (male vs. female) | 1.599 | 1.043 to 2.478 | 0.033* |
| Perioperative 5‐FU | 1.083 | 0.7028 to 1.663 | 0.7154 |
|
| 1.247 | 0.8153 to 1.911 | 0.3089 |
| Pathological TNM stage (Stage II vs. III) | 0.467 | 0.2820 to 0.7470 | 0.0021* |
| Tumor sidedness (left vs. right) | 1.13 | 0.7289 to 1.758 | 0.5862 |
| Tumor sidedness (not specified vs. right) | 1.743 | 0.5173 to 4.410 | 0.2968 |
|
| 0.1389 | 0.007847 to 0.6337 | 0.0508 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic factors in colorectal cancer · Colorectal Cancer Treatments and Studies · Cancer Genomics and Diagnostics
Background
1
Prognostication of early stage colorectal cancer (CRC) at initial diagnosis has traditionally relied on gross tumor anatomy and histology [1]. Due to the high risk of recurrence, the current standard of care treatment for Stage III CRC after surgical resection is adjuvant chemotherapy, whereas for Stage II CRC, due to the overall intermediate risk of recurrence, the decision for adjuvant chemotherapy is individualized based on clinicopathological risk factors such as the anatomy of the primary lesion or the presence of high‐risk histological features [2]. Only a handful of prognostic molecular biomarkers have been reliably utilized in clinical settings, such as CDX2, of which the loss of expression leads to inferior disease‐specific survival in Stages II and III colon cancer [3].
The mammalian rat sarcoma (RAS) gene family or paralogues, HRAS, KRAS, and NRAS, encode GTP‐binding hydrolases that act as a molecular switch of cellular signaling pathways. RAS proteins exist in either an “inactive” conformation when bound to GDP or “active” conformation when mitogenic signals catalyze exchange of the GDP to GTP, which activates a downstream signaling pathway and promotes cell growth. When the bound GTP is hydrolyzed to GDP, the RAS protein converts to its “inactive” state. Oncogenic RAS gene mutations result in increased affinity to GTP or loss of GTP hydrolysis [4]. Pathologic KRAS mutation is found in approximately 40% of CRC patients, of which approximately 85% occur in codons 12, 13, and 61 [5] and is associated with worse overall survival (OS). The mutation also demonstrates inferior response to standard of care first‐line 5‐fluorouracil (5‐FU)‐based treatments, as well as anti‐EGFR therapy, compared to those harboring wild‐type KRAS [6]. NRAS mutation is present in approximately 5% of colon cancers and is associated with poor survival outcomes. HRAS mutation is extremely rare in CRC, and its prognostic implication remains unclear to date [7, 8]. The prognostic implication of RAS paralogue gene activity at the transcript level is also obscure.
Interestingly, there is increasing evidence suggesting that different oncogenic RAS paralogues may associate with distinct biological phenotypes [9]. In a mouse model study, KRAS ^G12D^ promoted hyperproliferation, whereas NRAS ^G12D^ did not affect proliferation but instead suppressed apoptosis of colonic epithelium [10]. A comprehensive review of mutation profiles in advanced CRC patients demonstrated a significantly higher frequency of HRAS mutations in the high tumor mutation burden (TMB) tumors compared to low TMB tumors [11]. Furthermore, experimental evidence suggests that proto‐oncogene (or wild‐type) RAS signaling in cancer cells harboring an oncogenic RAS mutation also contributes to cellular proliferation that is independent from the coexisting oncogenic RAS mutation [12], which underscores the potential therapeutic implications of RAS proto‐oncogenes.
The aim of this study was to investigate the prognostic implications of HRAS in Stages II and III CRC and retrospectively analyzed the transcriptional expression profiles of the RAS family using the tissue RNA‐Seq database of CRC patients. The Cancer Genome Atlas (TCGA) is considered the largest and most comprehensive cancer genomic dataset available. Among the TCGA colorectal adenocarcinoma (COADREAD) cohort, 372 Stages II and III CRC cases with primary tumor RNA‐Seq and censorship data were accessible, which was deemed sub‐optimally small for the purpose of this study. In addition, the original publication of the TCGA‐COADREAD project dates back to 2012 [13] and may not reflect recent survival trends among CRC patients. Therefore, the clinical and RNA‐Seq data from Stages II and III CRC patients enrolled in the Oncology Research Information Exchange Network (ORIEN) AVATAR program was selected as the primary dataset for this study.
Methods
2
ORIEN is an ongoing multicenter collaboration between 19 cancer centers in the United States, in which cancer patients are prospectively enrolled in the Total Cancer Care protocol, approved by individual institutions' review board, launched in 2016. Patients with available tumor tissues for molecular profiling are included in the ORIEN AVATAR program. Clinicopathological, treatment, and outcome data are abstracted and periodically updated by individual sites. All molecular profiling and data management are maintained by Aster Insights in collaboration with all participating cancer centers. All clinical and molecular data are de‐identified and made available to investigators from participating institutions upon request and study approval.
From the ORIEN AVATAR cohort, 734 patients with Stages II and III CRC were identified as of April 16, 2024 (Table S1). Primary (n = 560) or metastatic (n = 174, if primary tumor specimen is not available) tumor tissue whole‐exome sequencing and RNA‐Seq data and their associated clinical data were retrieved. Combat‐Seq from the sva R package was used for transcript count batch correction [14]. Samples that belonged to a batch group with a single sample were excluded. Batch‐corrected transcript counts were normalized to transcripts per million (TPM).
Clinicopathological and batch‐corrected primary tumor tissue RNA‐Seq data (V2 RSEM) of 390 Stages II and III CRC patients enrolled in TCGA‐COADREAD Pan Cancer project were retrieved from the cBio cancer genomics portal [15]. Censorship information was available for 372 cases.
Log‐rank p‐value and hazard ratio (HR) were derived from Kaplan–Meier survival analysis. All statistical analyses were performed using GraphPad Prism software.
Results
3
Transcript levels of HRAS, KRAS, and NRAS in Stages II and III CRC were unimodally distributed (Figure 1a–c). The 3.7‐year HR for death in patients with “very high” or the “top 5%” HRAS transcript level (n = 24) compared to the rest (n = 360) was 0.15 (95% CI: 0.07–0.34, p = 0.03; Figure 1d). However, no significant differential HR was seen in patients with the top 5% KRAS (Figure 1e) or NRAS (Figure 1f) transcript levels. When patients with primary tumor RNA‐Seq data were analyzed exclusively, a similar trend towards improved 3.7‐year OS associated with “very high” HRAS transcript levels was observed, although it did not reach statistical significance (Figure S1a,d). Again, no significant differential HR was associated with “very high” KRAS or NRAS (Figure S1b,c,e,f).
Distribution of (a) HRAS, (b) KRAS, and (c) NRAS transcript levels in combined Stages II and III CRC. Gray vertical dashed lines represent the “very high” or the “top 5%” transcript level cutoffs. Corresponding Kaplan–Meier OS analysis at 3.7 years by (d) HRAS, (e) KRAS, and (f) NRAS transcript levels. ns, Not statistically significant.
To identify clinicopathological factors which may contribute to the superior OS observed in patients with “very high” HRAS transcript levels, Cox regression analysis was performed, which further revealed Stage II versus III with HR 0.47 (95% CI: 0.28–0.75, p = 0.002) and male versus female sex with HR 1.60 (95% CI: 1.04–2.48, p = 0.033) as significant prognostic parameters in this patient population (Table 1). “Very high” HRAS transcript levels trended towards better survival, although they did not reach statistical significance (HR 0.14, 95% CI: 0.01–0.63, p = 0.051). The “very high” HRAS transcript level‐associated superior OS was preserved within Stages II and III (Figure S2a,b) as well as within male and female sex (Figure S2c,d), respectively, although statistical significance was not reached, likely due to insufficient sample size. Notably, prior exposure to perioperative 5‐FU‐based chemotherapy was not identified as a significant parameter.
Interactions between the RAS family genes have been hypothesized and reported previously in an animal cancer model study [16]. To investigate whether KRAS transcript levels had any influence on the HRAS expression‐associated differential OS, Stages II and III CRC patients were dichotomized by KRAS transcript levels. To avoid confounding effects of underlying RAS pathway gene mutation on survival outcomes, patients with pathologic mutations in KRAS (codons 12, 13, and 61), NRAS (any exonic), or BRAF (any) were excluded. Of note, BRAF encodes for serine/threonine kinase that acts downstream of RAS paralogues, and its oncogenic mutations are prevalent in approximately 10% of CRC [17]. To optimize statistical power, the cohort median transcript level of HRAS and KRAS was used as a cutoff to define “high” versus “low” HRAS and KRAS transcript levels, respectively (Figure S3a). The 5‐year HR for death in patients with “high” HRAS transcript levels with concomitant “low” KRAS transcript levels (n = 77) compared to those with “high” KRAS transcript levels (n = 53) was 0.42 (95% CI: 0.18–0.96, p = 0.03; Figure S3b). On the other hand, “low” and “high” KRAS transcript levels did not differentiate HR for death in patients with “low” HRAS transcript levels (Figure S3c). These findings remained consistent even when the analysis was limited to patients with primary tumor RNA‐Seq data. The 5‐year HR for death in patients with “high” HRAS transcript levels with concomitant “low” KRAS transcript levels (n = 62) compared to those with “high” KRAS transcript levels (n = 48) was 0.22 (95% CI: 0.08–0.62, p = 0.004; Figure S4a,b). However, “low” and “high” KRAS transcript levels did not differentiate HR for death in patients with “low” HRAS transcript levels (Figure S4c).
When analyses were performed in patients harboring pathologic KRAS mutations (codons 12, 13, and 61) but not NRAS or BRAF mutations, “low” and “high” KRAS transcript levels no longer differentiated the 5‐year HR for death in patients with “low” and “high” HRAS transcript levels (Figure S5a–c). Again, these findings remained consistent even when the analysis was limited to patients with primary tumor RNA‐Seq data (Figure S6a–c).
Similar analyses were performed to investigate whether NRAS transcript levels had any influence on the HRAS expression‐associated differential OS, but “low” and “high” NRAS transcript levels did not differentiate the 5‐year HR for death in patients with “low” and “high” HRAS transcript levels (Figure S7a–c). Again, these findings remained consistent even when the analysis was limited to patients with primary tumor RNA‐Seq data (Figure S8a–c).
To further identify clinicopathological factors which may contribute to the superior 5‐year OS observed in Stages II and III CRC patients with “high” HRAS transcript levels with no pathologic KRAS, NRAS, or BRAF mutations, Cox regression analysis was performed, which revealed left versus right primary tumor sidedness with HR for death at 3.1 (95% CI: 1.13–10.01, p = 0.039) as a significant prognostic parameter in this subpopulation (Table S2). Subsequent Kaplan–Meier survival analysis confirmed that patients expressing “high” HRAS with concomitant “low” KRAS transcript levels were associated with superior OS compared to those with concomitant “high” KRAS transcript levels with HR for death at 0.17 (95% CI: 0.03–0.99, p = 0.06) when patients had right‐sided primary tumor (Figure S9a), but not when patients had left‐sided primary tumor (Figure S9b). When patients with primary tumor RNA‐Seq data were analyzed exclusively, a similar trend towards improved 5‐year OS associated with “high” HRAS with concomitant “low” KRAS was present that is more pronounced in patients with right‐sided primary tumor (Figure S10a), compared to those with left‐sided primary tumor (Figure S10b), although statistical significance was not reached.
To validate these findings in an independent dataset, the association between RAS transcript levels and OS was investigated in Stages II and III CRC patients with no pathologic KRAS, NRAS, or BRAF mutation in the TCGA‐COADREAD cohort, although analyses were limited due to small sample size. All RNA‐Seq tissue specimens were from the primary tumor. Again, transcript levels of HRAS, KRAS, and NRAS were unimodally distributed (Figure S11a–c). The 40‐month HR for death in patients with “very high” (defined as the “top 15%” due to smaller sample size) HRAS transcript level (n = 22) was not significantly different compared to the rest (n = 140), although there was a trend towards improved OS (Figure S11d), which is consistent with the observation from the ORIEN dataset. Similarly, no significant differential HR was seen in patients with the top 15% KRAS (Figure S11e) or NRAS transcript levels (Figure S11f). Next, the influence of KRAS transcript level on HRAS‐dependent OS was analyzed. Given skewness of the transcript level distributions, the cutoff to define “high” versus “low” KRAS transcript levels was set differently for “high” and “low” HRAS level groups (Figure S12a). The 40‐month HR for death in patients with “high” HRAS transcript levels and concomitant “low” KRAS transcript levels (n = 7) was not significantly different compared to those with “high” KRAS transcript levels (n = 68), but again there was a trend towards improved OS (Figure S12b). On the other hand, “low” and “high” KRAS transcript levels did not differentiate the HR for death in patients with “low” HRAS transcript levels (Figure S12c).
Discussion
4
Strikingly, in Stages II and III CRC, superior OS was associated with the subgroup of patients exhibiting high HRAS transcript levels compared to those expressing low HRAS transcript levels, which was most pronounced in patients with concomitant low KRAS transcript levels without pathologic KRAS mutation, with right‐sided primary tumor location. Approximately 24% of the RNA‐Seq data in the ORIEN dataset were from non‐primary tumor specimens, but these findings were consistently present even when primary tumor RNA‐Seq data was analyzed exclusively.
Contrary to the notion that RAS family genes are proto‐oncogenic, this study demonstrates that “high” HRAS transcript levels are associated with superior OS. The “high” HRAS‐associated OS benefit was most pronounced in patients with right‐sided primary tumors concomitantly expressing “low” KRAS transcript levels in the absence of pathologic KRAS mutations. When expressed at high levels, HRAS may counteract the proto‐oncogene KRAS but not the oncogene KRAS. Antagonistic interaction between the RAS paralogues has been previously reported in a KRAS‐mutated lung cancer mouse model study using the CRISPR/Cas9 gene editing system, which demonstrated that HRAS and NRAS suppress tumor proliferation by directly interacting with oncogenic KRAS at the protein level [16]. Our study suggests that the antagonistic relationship between RAS paralogue genes may be more prevalent than previously assumed.
There are limitations to this study. First, even though ORIEN is a multicentric cohort, the findings of this study are based on a single dataset. It is reassuring that the survival analyses using a much smaller available dataset (TCGA‐COADREAD) demonstrated a similar trend towards improved survival outcome in the patient group with “high” HRAS and “low” KRAS, although statistical significance was not reached. Second, this was a retrospective study. Further validation with an independent cohort dataset and prospective investigation is warranted.
Only a handful of gene expression level‐based prognostic biomarkers for CRC are reliably used in clinical settings. Loss of expression of mismatch repair (MMR) genes, namely MLH1, MSH2, MSH6, and PMS2, is associated with an excellent response to checkpoint inhibitor immunotherapies. Loss of expression of CDX2, which is a caudal‐type homeobox transcription factor, portends a higher risk of recurrence, which warrants adjuvant chemotherapy in Stage II CRC, especially when other recurrence risk factors are absent [3]. Overexpression of HRAS was previously associated with responsiveness to lenvatinib in human gastro‐entero‐pancreatic neuroendocrine tumor cell lines [18], but the prognostic implication of HRAS expression has not been reported in CRC to date. The potential of HRAS as a prognostic biomarker in Stages II and III CRC should be explored further.
Author Contributions
Donghyun Kim: conceptualization, writing – original draft, investigation, formal analysis, writing – review and editing, methodology, visualization. Saima Sharif: writing – review and editing, supervision. Juan Antonio Raygoza Garay: writing – review and editing, data curation. Avanish S. Bhakta: writing – review and editing, resources. Patrick M. Boland: writing – review and editing, resources. Michael J. Cavnar: writing – review and editing, resources. Michelle L. Churchman: writing – review and editing, project administration. Hassan Hatoum: writing – review and editing, resources. Lyen C. Huang: writing – review and editing, resources. Joseph Kim: writing – review and editing, resources. Richard Kim: writing – review and editing, resources. Robert W. Lentz: writing – review and editing, resources. Sarbajit Mukherjee: writing – review and editing, resources. Mary T. O'Donnell: writing – review and editing, resources. Benjamin Quartey: writing – review and editing, resources. Matthew J. Reilley: writing – review and editing, resources. Robert J. Rounbehler: writing – review and editing, project administration. Bodour Salhia: writing – review and editing, resources. Bryan P. Schneider: writing – review and editing, resources. Carlos H. Chan: writing – review and editing, supervision, formal analysis, resources, methodology, writing – original draft, conceptualization.
Ethics Statement
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Figure S1: Distribution of (a) HRAS, (b) KRAS, and (c) NRAS transcript levels in combined Stages II and III CRC patients, in primary tumor only. On Gray vertical dashed lines represent the “very high” or the “top 5%” transcript level cutoffs. Corresponding Kaplan–Meier OS analysis at 3.7 years by (d) HRAS, (e) KRAS, and (f) NRAS transcript levels. ns, not statistically significant.
Figure S2: Kaplan–Meier OS analysis at 3.7 years by HRAS transcript levels in (a) Stage II, (b) Stage III, (c) male‐only in Stages II and III combined, and (d) female‐only in Stages II and III combined CRC patient subgroups. ns, not statistically significant.
Figure S3: Kaplan–Meier OS analysis at 5 years by HRAS and KRAS transcript levels in Stages II and III CRC patients with no pathologic KRAS, NRAS, or BRAF mutations. (a) Scatter plot of HRAS and KRAS transcript levels; gray dashed lines represent median value, (b) Kaplan–Meier OS analysis in patients with “high” HRAS transcript levels by “low” versus “high” KRAS transcript levels, and (c) Kaplan–Meier OS analysis in patients with “low” HRAS transcript levels by “low” versus “high” KRAS transcript levels. ns, not statistically significant.
Figure S4: Kaplan–Meier OS analysis at 5 years by HRAS and KRAS transcript levels in Stages II and III CRC patients with no pathologic KRAS, NRAS, or BRAF mutations, in primary tumor only. (a) Scatter plot of HRAS and KRAS transcript levels; gray dashed lines represent median value, (b) Kaplan–Meier OS analysis in patients with “high” HRAS transcript levels by “low” versus “high” KRAS transcript levels, and (c) Kaplan–Meier OS analysis in patients with “low” HRAS transcript levels by “low” versus “high” KRAS transcript levels. ns, not statistically significant.
Figure S5: Kaplan–Meier OS analysis at 5 years by HRAS and KRAS transcript levels in Stages II and III CRC patients with pathologic KRAS mutation but without NRAS or BRAF mutations. (a) Scatter plot of HRAS and KRAS transcript levels; gray dashed lines represent median value, (b) Kaplan–Meier OS analysis in patients with “high” HRAS transcript levels by “low” versus “high” KRAS transcript levels, and (c) Kaplan–Meier OS analysis in patients with “low” HRAS transcript levels by “low” versus “high” KRAS transcript levels. ns, not statistically significant.
Figure S6: Kaplan–Meier OS analysis at 5 years by HRAS and KRAS transcript levels in Stages II and III CRC patients with pathologic KRAS mutation but without NRAS or BRAF mutations, in primary tumor only. (a) Scatter plot of HRAS and KRAS transcript levels; gray dashed lines represent median value, (b) Kaplan–Meier OS analysis in patients with “high” HRAS transcript levels by “low” versus “high” KRAS transcript levels, and (c) Kaplan–Meier OS analysis in patients with “low” HRAS transcript levels by “low” versus “high” KRAS transcript levels. ns, not statistically significant.
Figure S7: Kaplan–Meier OS analysis at 5 years by HRAS and NRAS transcript levels in Stages II and III CRC patients with no pathologic KRAS, NRAS, or BRAF mutations. (a) Scatter plot of HRAS and NRAS transcript levels; gray dashed lines represent median value, (b) Kaplan–Meier OS analysis in patients with “high” HRAS transcript levels by “low” versus “high” NRAS transcript levels, and (c) Kaplan–Meier OS analysis in patients with “low” HRAS transcript levels by “low” versus “high” NRAS transcript levels. ns, not statistically significant.
Figure S8: Kaplan–Meier OS analysis at 5 years by HRAS and NRAS transcript levels in Stages II and III CRC patients with no pathologic KRAS, NRAS, or BRAF mutations, in primary tumor only. (a) Scatter plot of HRAS and NRAS transcript levels; gray dashed lines represent median value, (b) Kaplan–Meier OS analysis in patients with “high” HRAS transcript levels by “low” versus “high” NRAS transcript levels, and (c) Kaplan–Meier OS analysis in patients with “low” HRAS transcript levels by “low” versus “high” NRAS transcript levels. ns, not statistically significant.
Figure S9: Kaplan–Meier OS analysis at 5 years in patients with high HRAS transcript levels by “low” versus “high” KRAS transcript levels with (a) right‐sided primary tumor and (b) left‐sided primary tumor in the absence of pathologic KRAS, NRAS and BRAF mutations. ns, not statistically significant.
Figure S10: Kaplan–Meier OS analysis at 5 years in patients with high HRAS transcript levels by “low” versus “high” KRAS transcript levels with (a) right‐sided primary tumor and (b) left‐sided primary tumor in the absence of pathologic KRAS, NRAS and BRAF mutations, in primary tumor only. ns, not statistically significant.
Figure S11: Distribution of (a) HRAS, (b) KRAS, and (c) NRAS transcript levels in combined Stages II and III CRC of TCGA‐COADREAD cohort. Gray vertical dashed lines represent the “very high” or the “top 15%” transcript level cutoffs. Corresponding Kaplan–Meier OS analysis at 40 months by (d) HRAS, (e) KRAS, and (f) NRAS transcript levels. ns, not statistically significant.
Figure S12: Kaplan–Meier OS analysis at 40 months by HRAS and KRAS transcript levels in Stages II and III CRC patients of TCGA‐COADREAD cohort with no pathologic KRAS, NRAS, or BRAF mutations. (a) Scatter plot of HRAS and KRAS transcript levels; gray vertical dashed line represents median value, and gray horizontal dashed lines represent selected cutoffs, (b) Kaplan–Meier OS analysis in patients with “high” HRAS transcript levels by “low” versus “high” KRAS transcript levels, and (c) Kaplan–Meier OS analysis in patients with “low” HRAS transcript levels by “low” versus “high” KRAS transcript levels. ns, not statistically significant.
Table S1: ORIEN AVATAR Stages II and III CRC patient summary.
Table S2: Cox regression analysis on overall survival of Stages II and III colorectal cancer with high HRAS transcript expression in KRAS(−); BRAF(−); NRAS(−) genotypic background at 5 years.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1M. B. Amin , S. B. Edge , F. L. Greene , et al., AJCC Cancer Staging Manual (Springer International Publishing, 2018).
- 2A. B. Benson, 3rd , D. Schrag , M. R. Somerfield , et al., “American Society of Clinical Oncology Recommendations on Adjuvant Chemotherapy for Stage II Colon Cancer,” Journal of Clinical Oncology 22, no. 16 (2004): 3408–3419.15199089 10.1200/JCO.2004.05.063 · doi ↗ · pubmed ↗
- 3P. Dalerba , D. Sahoo , S. Paik , et al., “CDX 2 as a Prognostic Biomarker in Stage II and Stage III Colon Cancer,” New England Journal of Medicine 374, no. 3 (2016): 211–222.26789870 10.1056/NEJ Moa 1506597 PMC 4784450 · doi ↗ · pubmed ↗
- 4C. Johnson , D. L. Burkhart , and K. M. Haigis , “Classification of KRAS‐Activating Mutations and the Implications for Therapeutic Intervention,” Cancer Discovery 12, no. 4 (2022): 913–923.35373279 10.1158/2159-8290.CD-22-0035 PMC 8988514 · doi ↗ · pubmed ↗
- 5R. Dienstmann , K. Connor , A. T. Byrne , and C. Consortium , “Precision Therapy in RAS Mutant Colorectal Cancer,” Gastroenterology 158, no. 4 (2020): 806–811.31972237 10.1053/j.gastro.2019.12.051 · doi ↗ · pubmed ↗
- 6A. R. Moore , S. C. Rosenberg , F. Mc Cormick , and S. Malek , “RAS‐Targeted Therapies: Is the Undruggable Drugged?,” Nature Reviews Drug Discovery 19, no. 8 (2020): 533–552.32528145 10.1038/s 41573-020-0068-6PMC 7809886 · doi ↗ · pubmed ↗
- 7J. M. Loree , Y. Wang , M. A. Syed , et al., “Clinical and Functional Characterization of Atypical KRAS/NRAS Mutations in Metastatic Colorectal Cancer,” Clinical Cancer Research 27, no. 16 (2021): 4587–4598.34117033 10.1158/1078-0432.CCR-21-0180 PMC 8364867 · doi ↗ · pubmed ↗
- 8I. A. Prior , P. D. Lewis , and C. Mattos , “A Comprehensive Survey of Ras Mutations in Cancer,” Cancer Research 72, no. 10 (2012): 2457–2467.22589270 10.1158/0008-5472.CAN-11-2612 PMC 3354961 · doi ↗ · pubmed ↗