Research progress of PYK2 in digestive system diseases
Yiyao Duan, Mingzhu Xie, Hui Wang, Sijing Chen, Jun Hu, Xujia Chen, Hong Ping Jia, Ningyan Zhang, Ling Peng, Xiang Li, Hameed Ullah Khan, Die Hu, Rong Qin

TL;DR
This review explores the role of PYK2 in digestive system diseases, particularly its involvement in cancer progression and potential as a drug target.
Contribution
The paper provides a comprehensive analysis of PYK2's role in gastrointestinal diseases and its therapeutic potential.
Findings
PYK2 is involved in tumor cell adhesion, growth, and metastasis in digestive system cancers.
PYK2 inhibitors show therapeutic potential for improving cancer management and prognosis.
PYK2 contributes to tumor progression through specific signaling pathways.
Abstract
Belonging to the focal adhesion kinase (FAK) family, proline-rich tyrosine kinase 2 (PYK2) is a non-receptor tyrosine kinase, has become a focal point in cancer research owing to its essential participation in the formation and dissemination of tumors. Studies have shown that this kinase controls various cellular activities, including: tumor cell adhesion, growth, multiplication, specialization, and detachment, making it a promising target for developing anticancer drugs. The goal of this review is to analyze the multifaceted role of PYK2 in gastrointestinal disease, focusing on its contribution to tumor progression, associated signaling pathways, and the therapeutic potential of PYK2 inhibitors in improving disease management and prognosis.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
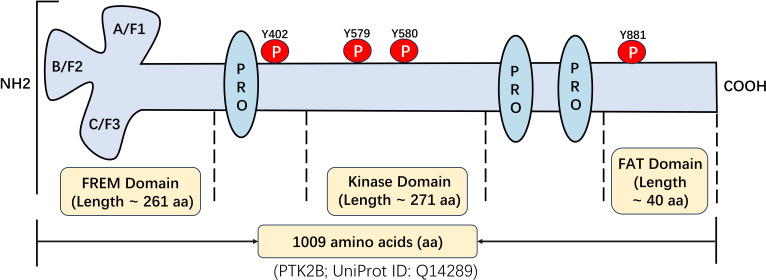 Figure 1
Figure 1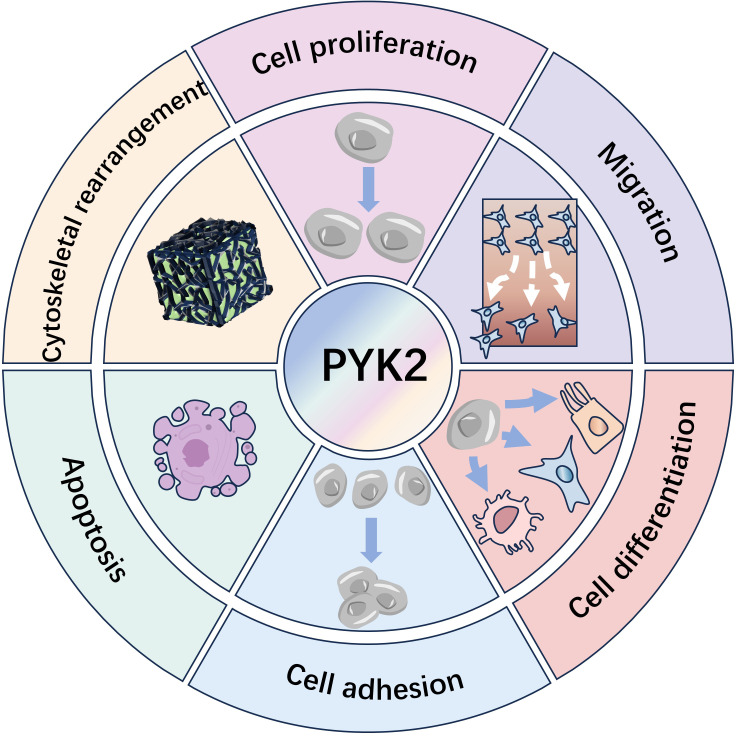 Figure 2
Figure 2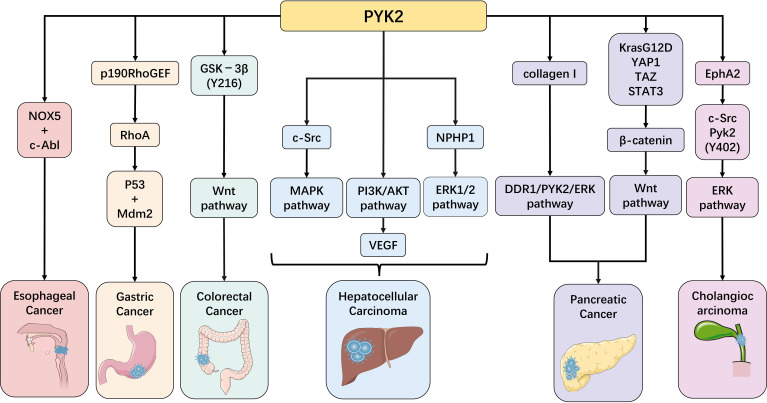 Figure 3
Figure 3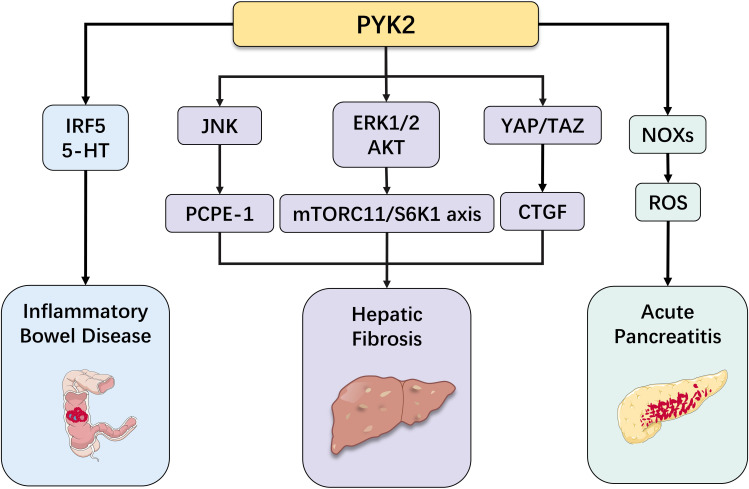 Figure 4
Figure 4| Cancer Type | Alteration Frequency | Common Alterations | Clinical Association | References |
|---|---|---|---|---|
| ESCC | ~10-15% | Amplification | Lymph node metastasis | ( |
| GC | 5-8% | Missense mutations (Y402) | Advanced TNM stage; | ( |
| CRC | 12-18% | Amplification | Liver metastasis; | ( |
| HCC | ~20% | Focal gains (8p21.1) | Vascular invasion | ( |
| PDAC | 15-20% | Amplification | Stromal remodeling; | ( |
| IBD(UC) | GWAS locus | SNP: rs4750316 | Disease severity; CD11c+ macrophage infiltration | ( |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCell Adhesion Molecules Research · Angiogenesis and VEGF in Cancer · Cancer Cells and Metastasis
Introduction
1
Part of the FAK family, PYK2 is referred to as cellular adhesion kinase β (CAK-β), which is mainly distributed in the cytoplasm, and thus belongs to the Cytoplasmic tyrosine kinase (1–3). This enzyme belongs to a unique group of protein kinases that target tyrosine residues on proteins for phosphorylation. Various cell types and tissues universally express PYK2, such as neural tissues, endothelial cells, brain cells, fibroblasts, platelets, and specific hematopoietic cells (4–6).
Structure and function of PYK2
2
Structural domains of PYK2
2.1
PYK2 was initially cloned and identified in 1995 as a gene encoding a protein tyrosine kinase. This gene is found on chromosome 8p21.1 within the human genome, featuring a cDNA sequence that spans 4,048 base pairs and with a molecular mass of ~116 kDa (7). Structural analysis reveals that PYK2 shares significant homology with FAK, exhibiting 46% identity and 65% similarity at the protein level (8). PYK2 contains three core functional domains (9): (1) the FERM (4.1 protein, Ezrin, Radixin, Moesin) domain at the N-terminal: structurally, the FERM domain adopts a compact cloverleaf conformation composed of three distinct structural modules (Figure 1). These modules are recognized for their role in facilitating protein-protein and protein-lipid interactions (10); (2) the central domain responsible for tyrosine kinase activity: it contains catalytic residues (Y579/Y580 in the activation loop) and connects to FERM via a conserved linker harboring Y402—a scaffold site for phosphorylation-dependent signaling; and (3) the C-terminal FAT (Focal Adhesion Targeting) domain targets focal adhesions and features the Y881 phosphorylation site, mediates focal adhesion targeting. The spatial organization of these domains determines the function of PYK2 as a signal transduction hub and scaffold.
Structural and functional characterization of PYK2: identification of key domains and post-translational modification sites.
The primary PYK2 splice variant, PYK2-M, lacks kinase activity due to C-terminal truncation but retains the FERM domain, enabling it to function as a dominant-negative scaffold (11, 12). By competitively binding shared interactors (e.g., integrins, IRF5), PYK2-M disrupts PYK2-H-dependent signaling, suppressing cell migration and pro-inflammatory transcription (13). In digestive pathologies—where PYK2-H drives cancer metastasis (HCC/PDAC) and inflammation (IBD)—PYK2-M may similarly antagonize these processes through kinase-independent scaffolding, though direct evidence remains limited and warrants investigation.
Nuclear functions
2.2
Nuclear localization and nucleocytoplasmic shuttling mechanisms of PYK2
2.2.1
The nuclear functions of PYK2 in gastrointestinal tumors are regulated through a multifaceted mechanism involving a classical nuclear localization signal (NLS) located in the F2 subdomain of its FERM domain and a nuclear export signal (NES) embedded within its kinase domain (14). These signals coordinately govern the nucleocytoplasmic shuttling of PYK2. In gastrointestinal tumor cells, calcium influx or mechanical stress activates calcineurin. Activated calcineurin dephosphorylates PYK2 at Ser778, which impairs the function of the NES, leading to enhanced nuclear accumulation of PYK2 (15). For instance, in pancreatic ductal adenocarcinoma (PDAC), increased matrix stiffness induces Piezo1-mediated calcium influx, triggering PYK2 nuclear translocation (16). Furthermore, SUMOylation enhances PYK2 nuclear retention, whereas ubiquitination facilitates its nuclear export. Supporting this, SUMOylation-deficient PYK2 mutants exhibit significantly impaired nuclear accumulation in PDAC models, underscoring the critical role of post-translational modifications in PYK2 nucleocytoplasmic trafficking (17).
Nuclear scaffolding functions of PYK2
2.2.2
As a nuclear scaffolding protein, PYK2 remodels transcriptional networks through multiple mechanisms, including transcription factor cooperativity, chromatin remodeling and epigenetic regulation, and assembly of nuclear signaling complexes, thereby driving the malignant phenotypes of gastrointestinal tumors (18). (1) Transcription Factor Cooperativity: PYK2 directly interacts with GATA-binding protein 4 (GATA4), facilitating its binding to the cyclin D1 promoter and accelerating cell cycle progression (19). In gastric cancer cells, the PYK2-GATA4 complex upregulates c-Myc expression, enhancing tumor cell proliferation.
Additionally, PYK2 interacts with CREB (cAMP response element-binding protein), activating the transcription of inflammatory cytokines such as IL-17A, which contributes to tumor microenvironment remodeling (20, 21). (2) Chromatin Remodeling and Epigenetic Regulation: Notably, in hepatocellular carcinoma models, dissociation of the PYK2-MBD2 complex is associated with activation of Wnt pathway-related genes (e.g., Axin2), promoting tumor cell invasion (22, 23). (3) Assembly of Nuclear Signaling Complexes: PYK2 recruits Src-family kinases (SFKs) in the nucleus, forming a PYK2-Src-Gab1 signaling module that activates the PI3K-AKT pathway (24). In esophageal cancer, this complex enhances the nuclear activity of YAP/TAZ, promoting cancer stem cell maintenance (25). These diverse nuclear scaffolding functions highlight how PYK2, upon nuclear translocation, acts as a central hub for rewiring transcriptional programs to promote gastrointestinal tumorigenesis.
Activation mechanisms of PYK2
2.3
Y402 phosphorylation serves as a master switch for PYK2 activation. Diverse stimuli converge on this event through five primary mechanisms:
Cell adhesion-mediated activation
2.3.1
Integrin engagement (e.g., with fibronectin) induces FAK-mediated phosphorylation of Y402. Phosphorylated Y402 recruits Src via its SH2 domain, leading to Src-mediated phosphorylation of Y579/Y580 in the activation loop and full kinase activation (26).
Ca²+ and PKC-dependent activation
2.3.2
This mechanism is commonly seen in vascular smooth muscle depolarization or neuronal signal transduction. For example, depolarization of vascular smooth muscle triggers the inflow of Ca2 +, activates PKC, and triggers the above-mentioned core activation pathway (27).
Regulatory role of the FERM domain
2.3.3
Under resting conditions, the FERM domain forms a β-sheet interaction with the linker region of PYK2, occluding the Y402 site. Disruption of this interaction (e.g., via the K60P mutation) exposes Y402, enhancing phosphorylation (28).
Oxidative stress-induced activation
2.3.4
Under oxidative stress (e.g., H_2_O_2_ stimulation), phospholipase D2 (PLD2) activation leads to Y402 phosphorylation, subsequently activating PI3K/Akt pathway to exert a cell-protective effect (29).
Through the above-mentioned mechanism, the activated (phosphorylated) PYK2 serves as a critical regulator, coordinating essential cellular activities. These include cytoskeletal remodeling, adhesion signaling, proliferation, motility, phenotypic differentiation, apoptosis modulation, and transcriptional regulation. (Figure 2) (7, 30).
PYK2: A comprehensive overview of its established biological roles.
Regulatory mechanisms governing PYK2 expression and activity
2.4
Post-transcriptional regulation
2.4.1
Regarding the post-transcriptional regulation of PYK2, it is mainly through microRNA-mediated expression regulation. Various microRNAs (miRNAs) inhibit the post-transcriptional expression of PYK2 by binding to the 3’ untranslated region (3’UTR) of PYK2’s mRNA to regulate cancer progression (31–33). For example, in hepatocellular carcinoma (HCC), miR-23b directly targets the 3’UTR of PYK2 and reduces the level of PYK2 protein, thereby inhibiting epithelial-mesenchymal transition (EMT) and tumor metastasis. Mechanistically, miR-23b reduces the expression of matrix metalloproteinases (MMPs) by weakening the PYK2-mediated AKT/mTOR signaling pathway and blocks the invasive ability of cancer cells (31). MiR-214 inhibits PYK2 expression by targeting its mRNA, thereby blocking the PI3K/AKT pathway and inhibiting cell proliferation (32). Down-regulation of miR-517a and miR-517c alleviates their inhibitory effect on PYK2, leading to elevated PYK2 expression. Subsequently, PYK2 promotes cell proliferation by activating ERK1/2 signaling (33).
Post-translational modifications
2.4.2
Post-translational modifications of PYK2 primarily include phosphorylation at Y402, as well as at Y654, which promotes nuclear translocation of β-catenin, which activates the Wnt signaling pathway and drives reprogramming of pancreatic acinar cells and tumor maintenance. This modification relieves ubiquitination degradation of β-catenin and prolongs its half-life (34). In addition to phosphorylation, the activity of PYK2 may also be regulated by ubiquitination (35). Phosphorylated PYK2 may be more readily recognized and ubiquitinated by Cbl-b, forming a “phosphorylation-ubiquitination” cascade that promotes its degradation.
Feedback loops
2.4.3
PYK2 is also involved in the cross-regulation of signaling networks. For example, in PDAC, PYK2 activates the Wnt/β-catenin pathway after phosphorylating β-catenin (Y654), inducing the expression of downstream genes (e.g., c-Myc, Cyclin D1) to promote cell proliferation. Meanwhile, the activation of Wnt signaling can upregulate the expression of PYK2, forming the “PYK2→β-catenin→Wnt→PYK2” feedback, driving the malignant transformation of precancerous cells (34). In addition, in breast cancer, PYK2 activates STAT3 (Y705 site), forming “PYK2→STAT3→PYK2” positive feedback (36). Although this loop was identified in breast cancer, similar mechanisms may operate in digestive tumors given STAT3’s established oncogenic role in PC (37).
These findings collectively demonstrate that PYK2 participates in the cross-regulation of multiple signaling pathways through the formation of positive feedback loops, and thus plays a key role in tumorigenesis and development.
Context-dependent PYK2 function: tissue, microenvironment, epigenetics
2.5
Context-dependent function refers to PYK2’s ability to act as either an oncogene or tumor suppressor based on tissue-specific cues, microenvironmental signals, and epigenetic regulation. The paradoxical functions of PYK2 as either an oncogene or tumor suppressor are principally governed by three interconnected determinants: (1) Tissue-specific interacting partners—in GC, PYK2 forms a nuclear complex with p53 and Mdm2 to drive p53 ubiquitination and degradation, thereby disabling a critical tumor-suppressive checkpoint (38–40). Conversely, in HCC, PYK2 physically associates with c-Src to activate MAPK signaling, directly promoting proliferation and invasion (41). (2) Tumor microenvironmental cues—hypoxia in ESCC induces Piezo1-mediated calcium influx, triggering PYK2 phosphorylation at Y402 and subsequent NOX5/c-Abl complex assembly to fuel tumor progression (16, 42); whereas in GC, mechanical stress from peristalsis may modulate RhoA-ROCK signaling through PYK2-p190RhoGEF crosstalk, potentially influencing cell detachment dynamics (43, 44). (3) Epigenetic regulation—miR-23b-mediated PYK2 suppression in HCC inhibits EMT and metastasis (31), whereas downregulation of miR-517a/c in the same malignancy elevates PYK2 expression to activate ERK1/2-driven proliferation (33). This functional plasticity necessitates context-aware therapeutic strategies: PYK2 inhibition may be beneficial in cancers where it acts as an oncogenic scaffold (e.g., ESCC, HCC, PDAC), but could prove detrimental in GC where its tumor-suppressive functions are compromised by downregulation.
Research progress on the role of PYK2 in digestive system diseases
3
In recent years, with the deepening of PYK2 research, its crucial role in digestive system diseases has become increasingly prominent. Particularly, the regulatory mechanisms of PYK2 in inflammatory diseases and tumorigenesis have emerged as a research hotspot in this field. This review will systematically summarize the latest research progress by focusing on the pivotal functions of PYK2 in various digestive system disorders, including inflammatory bowel disease, hepatic fibrosis, and gastrointestinal tumors (Figures 3, 4). These comprehensive analyses aim to provide novel theoretical foundations and potential intervention strategies for early diagnosis, targeted therapy, and prognosis improvement of related diseases.
PYK2 in gastrointestinal malignancies: implications in esophageal, gastric, colorectal, hepatic, gallbladder, and pancreatic carcinomas.
Role of PYK2 in other diseases of the digestive system (inflammatory bowel disease, hepatic fibrosis, acute pancreatitis).
The role of PYK2 in digestive system tumors
3.1
Esophageal cancer
3.1.1
EC, a common and lethal type of cancer worldwide, constitutes a serious risk to human health. There are two main histological subtypes: esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC), with ESCC being the most frequent (45). Current therapeutic strategies encompass surgery, radiotherapy, chemotherapy, and endoscopic interventions. Given its highly invasive and metastatic nature, the identification of potential prognostic biomarkers and therapeutic targets is of paramount importance. Current investigations have shown a significant elevation in nicotinamide adenine dinucleotide phosphate oxidase 5 (NOX5) expression levels in ESCC. Mechanistically, hypoxia has been shown to increase intracellular Ca2+ levels, thereby inducing phosphorylation of PYK2 at the Y402 site. This post-translational modification facilitates the interaction between PYK2 and NOX5, leading to the formation of a membrane-associated complex. Furthermore, PYK2 recruits c-Abl, which enhances the activity of NOX5 within this complex, ultimately promoting ESCC progression (42). Although PYK2 does not directly participate in the pathogenesis of EC, it plays a pivotal role in ESCC progression as a scaffold protein that facilitates c-Abl-mediated activation of NOX5 within the PYK2/NOX5 complex. Cisplatin, a commonly used chemotherapeutic agent in adjuvant therapy following radical esophagectomy, has been shown to have its efficacy influenced by PYK2. Moreover, PYK2 expression levels have been correlated with EC prognosis (46). In conclusion, PYK2 critically regulates both tumorigenesis and progression in EC. T Incorporating PYK2 inhibitors into postoperative adjuvant therapy could be a viable strategy for treating EC patients. Additional studies are needed to clarify the exact molecular mechanisms and assess the clinical effectiveness of therapies targeting PYK2.
Gastric carcinoma
3.1.2
Globally, GC represents the fifth most frequently diagnosed malignancy worldwide and ranks among the principal contributors to cancer-associated mortality (47). Elucidating the functional interplay between PYK2 and GC could provide important understanding of the molecular processes involved in disease progression and the creation of new treatments. Notably, studies have demonstrated a marked downregulation of PYK2 expression in GC tissues. Furthermore, clinical evidence has revealed an inverse correlation between the progressive decline in PYK2 expression levels and the advancement of TNM staging, strongly suggesting that function as a tumor suppressor during gastric carcinogenesis (38). The tumor suppressor p53 is a critical guardian of the genome, instrumental in inhibiting GC cell proliferation by inducing cell cycle arrest (primarily at G1/S checkpoint) and apoptosis in response to stress signals (48). Crucially, PYK2 directly antagonizes p53 function, particularly through the formation of a nuclear complex. As detailed in the “Nuclear Functions” section (Section 2.2), the N-terminal FERM domain of PYK2 binds p53 and recruits the E3 ubiquitin ligase Mdm2. This nuclear PYK2-p53-Mdm2 complex facilitates K48-linked polyubiquitination of p53, targeting it for proteasomal degradation (40). The degradation of nuclear p53 by this PYK2-mediated mechanism represents a key pathological event in GC. By reducing functional p53 levels, PYK2 effectively compromises p53-dependent cell cycle arrest, allowing dysregulated proliferation and contributing to tumor progression. This mechanism provides a molecular explanation for the observed inverse correlation between PYK2 expression and GC stage, linking PYK2 downregulation to the loss of a critical tumor suppressor checkpoint. PYK2 may also contributes to tumor metastasis, particularly in the initial detachment phase of cells. Previous studies have established that PYK2 can influence cancer progression by enhancing cell separation, as evidenced by its glucocorticoid-induced upregulation promoting osteoclast detachment (39). In the digestive system, trypsin, a digestive enzyme abundantly present in the stomach, small intestine, and colon, is commonly utilized for cell detachment. Trypsin causes cells to detach by degrading PYK2 via the ubiquitin-lysosome system, but PYK2 can counteract this detachment, though the exact regulatory processes are not yet fully understood (35). Accumulating experimental data indicate that mechanical forces generated by gastric peristalsis could potentially stimulate proliferation in both primary and metastatic GC cell lines, with concomitant modulation of proliferating cell nuclear antigen and p53 expression profiles (43). The tumor suppressor p53 is known to be instrumental in inhibiting the proliferation of GC cells (48). Interestingly, the N-terminal FERM domain of PYK2 can form a complex with p53 and Murine double minute-2 to regulate p53 levels (40). Furthermore, gastrointestinal motility is associated with the RhoA-ROCK signaling pathway, which plays a pivotal role in regulating cancer cell motility and invasion (49). In FAK−/− fibroblasts, PYK2 has been shown to promote RhoA activation and migration through enhanced p190RhoGEF expression (44). Collectively, these findings indicate a tumor-suppressive role for PYK2 in GC. Its downregulation facilitates tumor progression by impairing p53-mediated cell cycle arrest and genomic stability through nuclear complex formation and degradation, and potentially modulating cell detachment and motility pathways. Further investigation into the molecular mechanisms underlying PYK2’s role in GC progression, especially its nuclear scaffolding functions impacting cell cycle regulators beyond p53, may provide novel therapeutic targets for this devastating disease.
Colorectal cancer
3.1.3
CRC, a highly common malignancy globally, is affected by both environmental and genetic factors. Despite groundbreaking developments in both diagnostic technologies and treatment modalities since the turn of the century, there remain unexplored areas that necessitate further investigation. The progression of CRC is a complex, multifactorial process involving PYK2, which contributes not only to colonic smooth muscle contraction but also to intestinal homeostasis, epithelial repair, and tumorigenesis (50, 51). Canonical Wnt signaling constitutes a master regulatory pathway driving colorectal cancer development (52). Mechanistic studies have revealed that elevated FAK/PYK2 levels lead to dephosphorylation of glycogen synthase kinase-3β (GSK-3β) at the Y216 site, facilitating the recruitment of the ubiquitin ligase β-transducin repeats containing proteins (β-TrCP). Ubiquitination of β-catenin through this process activates Wnt signaling (53). These findings suggest that CRC progression is mediated through the PYK2/GSK-3β(Y216)/β-catenin regulatory axis, ultimately influencing cellular metabolism and contributing to malignant transformation. Furthermore, emerging evidence indicates that PYK2 may suppress CRC tumor growth by inhibiting mitochondrial oxidative phosphorylation (OXPHOS) in CRC cells (54). Notably, PYK2 has been identified as a novel independent prognostic marker for colon adenocarcinoma progression following surgical resection (55), with its overexpression demonstrating enhanced cellular proliferation and invasive capabilities. In conclusion, PYK2 critically regulates CRC development and disease outcomes. PYK2’s multifaceted pathway involvement establishes its druggability, warranting further mechanistic and clinical studies for CRC.
Hepatocellular carcinoma
3.1.4
Globally, HCC is the leading type of primary liver cancer. Notably, PYK2 overexpression has been detected in approximately 60% of HCC patients, where it not only promotes cancer cell proliferation but also enhances tumor cell invasiveness (56). Mechanistic investigations have revealed that PYK2 overexpression facilitates its binding with c-Src, forming a PYK2-c-Src signaling complex that undergoes autophosphorylation at Y402. This process activates c-Src, subsequently triggering multiple signaling cascades that activate the MAPK pathway, thereby promoting HCC cell proliferation and invasion (41). Furthermore, TMEM237 overexpression, frequently observed in HCC, has been shown to enhance HCC cell multiplication, movement, invasion, and epithelial-mesenchymal transition (EMT), correlating with poor clinical outcomes. Under hypoxic conditions, the transcriptional activity of the TMEM237 promoter is significantly enhanced through direct binding of HIF-1α (57). The TMEM237-NPHP1 association warrants special attention, as prior research has confirmed NPHP1’s ability to interact with PYK2, resulting in PYK2 phosphorylation and subsequent initiation of the PYK2-dependent ERK1/2 signaling cascade within renal tubular epithelial cells (58). Consequently, when TMEM237 is overexpressed, the interaction between NPHP1 and PYK2 is enhanced, forming a ternary complex that activates the PYK2/ERK1/2 pathway in HCC cells, thereby promoting tumor progression (57). PYK2’s involvement in tumor metastasis has been extensively documented. Researchers have proposed that the activation of the PI3K/AKT pathway by PYK2 enhances VEGF expression in HCC, facilitating peritumoral angiogenesis and inducing tumor metastasis (59). Alternatively, others suggest that PYK2 may enhance metastatic potential by modulating Rac1/RhoA activity to induce EMT (60). independent validation comes from Cao’s work showing miR-23b-mediated PYK2 downregulation effectively curbs EMT-driven HCC invasion (31). Additionally, both miR-214 overexpression and PTK2b/PYK2 knockdown triggered G1-phase arrest, effectively inhibiting malignant cell proliferation through cell cycle disruption (32). Both miR-517a and miR-517c inhibit G2/M phase transition and reduce mitotic activity, at least in part through direct targeting of PYK2, a key regulator of mitotic entry (33). In conclusion, PYK2 is critically involved in HCC progression and metastasis, functioning as both a predictive biomarker for platinum-based chemotherapy resistance and a potential therapeutic target. Further research into PYK2’s molecular mechanisms and its clinical applications may provide novel insights into HCC management strategies.
Pancreatic cancer
3.1.5
Despite notable progress in modern medicine, PC continues to be a highly fatal gastrointestinal cancer, with early detection remaining a significant challenge (61). Pancreatic ductal adenocarcinoma (PDAC) progresses through a multistep mechanism and constitutes the majority of pancreatic cancer cases. This process involves the reprogramming of precancerous acinar cells in response to various stimuli, including Kras oncogene mutations and pancreatitis, leading to pancreatic intraepithelial neoplasia and ultimately progressing to PDAC. During this transformation, alterations in PYK2 and other factors play crucial roles. Mechanistically, Yes-associated protein 1 (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), effectors of the Hippo pathway, regulate PYK2 transcription via STAT3 mediation. Subsequently, PYK2-mediated phosphorylation of β-catenin drives Wnt pathway activation, establishing PYK2 as a key orchestrator of PDAC tumorigenesis and progression (34, 37, 62). Critically, nuclear-translocated PYK2 acts as a scaffold to potentiate YAP/TAZ transcriptional activity. As detailed in Section 2.2.2, PYK2 recruits Src-family kinases (SFKs) within the nucleus to form a PYK2-Src-YAP/TAZ complex. This complex enhances the nuclear retention and transcriptional output of YAP/TAZ, directly activating genes essential for cancer stem cell maintenance (e.g., SOX9, OCT4) and EMT progression (18, 25). Concurrently, PYK2-phosphorylated β-catenin (pY654) translocates to the nucleus, where it displaces transcriptional repressors (e.g., HDAC1) from TCF/LEF-binding sites, further amplifying Wnt target genes (e.g., c-MYC, CYCD1) (34). This synergistic nuclear crosstalk between PYK2-YAP/TAZ and PYK2-β-catenin axes creates a self-reinforcing transcriptional circuit that drives PDAC aggressiveness. The importance of PYK2 in the development of PDAC is emphasized by these findings. A hallmark of PDAC is its extensive desmoplastic reaction, characterized by substantial type I collagen deposition. This collagen-rich microenvironment signals through integrins and discoidin domain receptor 1 (DDR1), with the DDR1b isoform interacting with PYK2 via Sch1. This interaction induces increased N-cadherin expression and facilitates EMT in PC cells, contributing to PDAC’s tumorigenic properties. During this process, collagen-mediated DDR1 activation induces PYK2-associated signaling pathways, potentially driving collagen-induced tumor progression (63, 64). Furthermore, Recent findings indicate that autophagy levels are crucial in the development and treatment of PDAC (65). Type I collagen activates the DDR1/PYK2/ERK signaling pathway, which primarily mediates autophagosome-lysosome fusion. The use of SH2 super binder to inhibit PDAC cell autophagy through DDR1 regulation has demonstrated antitumor effects (66). In conclusion, PYK2 demonstrates significant associations with pancreatic disease progression. The modulation of PYK2 expression and function holds promise for attenuating or reversing pancreatic pathological processes. Future investigations should prioritize elucidating the molecular mechanisms underlying PYK2’s role in pancreatic pathophysiology and developing targeted therapeutic interventions. These research endeavors may yield significant advancements in the clinical management of pancreatic disorders.
Cholangiocarcinoma
3.1.6
The detection rate of CCA - a biologically aggressive malignancy originating from biliary tract epithelia - has increased significantly with the advent of advanced diagnostic technologies. Despite this progress, research investigating the role of PYK2 in CCA remains limited. A notable study by Cui et al. (67) revealed that in CCA, PYK2 is activated by overexpressed Eph receptor A2 (EphA2). This activation subsequently triggers the PYK2/c-Src signaling pathway, which independently activates the ERK signaling cascade through a Raf/MEK-independent mechanism, ultimately modulating tumor cell invasion and metastatic potential.
The role of Pyk2 in other diseases of the digestive system
3.2
Inflammatory bowel disease
3.2.1
IBD, including the two principal phenotypes - ulcerative colitis (UC) and Crohn’s disease (CD), characterized by relapsing-remitting mucosal inflammation (68). Numerous genetic loci play significant roles in IBD pathogenesis, with PYK2 being identified as a nominated pathogenic gene for UC (69). PYK2 not only directly regulates UC progression but also functions as a regulatory factor in its pathological development. Interferon regulatory factor 5 (IRF5), a key immune response regulator, significantly contributes to the development of intestinal inflammation (70). PYK2 exacerbates intestinal inflammation through IRF5 phosphorylation (Tyr-171 in mice/Tyr-172 in humans), a modification that induces pathogenic CD11c+ macrophage accumulation in the inflamed colon (13, 71, 72). Furthermore, studies have demonstrated elevated levels of peripheral serotonin (5-hydroxytryptamine or 5-HT) in patients with intestinal inflammation (73). As an immunomodulatory neuroendocrine peptide, 5-HT not only regulates intestinal homeostasis and tumor biological processes (74, 75), but also induces B cell transformation into regulatory B cells (Bregs), enhancing their capacity to suppress intestinal inflammation (76). PYK2 influences IBD progression through its involvement in 5-HT secretion: upon binding with invariant natural killer T cells, enterochromaffin cells selectively sense lipid antigens, leading to CD1d Tyr332 recruitment and PYK2 activation. Subsequently, PYK2 regulates Kv1.2 channels through tyrosine phosphorylation, triggering Ca2+ influx and subsequent 5-HT release (77).
Hepatic fibrosis
3.2.2
PYK2, a critical regulator of cellular processes, has recently been identified as closely associated with liver health, particularly playing significant roles in lipid metabolism, inflammatory responses, and liver cirrhosis. Its aberrant activity may disrupt lipid homeostasis, exacerbate hepatic inflammation, promote liver fibrosis, and accelerate the progression of cirrhosis. Therefore, in-depth investigation of PYK2’s role in these pathways may provide new hope for developing targeted therapies for liver diseases. Specifically, PYK2 can activate the ERK1/2 and AKT-mediated mTORC1/S6K1 axis, enhancing lipogenesis and leading to hepatic steatosis (78). Furthermore, metabolic dysfunction-associated steatohepatitis is crucial in the development of liver steatosis., characterized by progressive extracellular matrix deposition and sterile hepatic inflammation, potentially culminating in end-stage liver complications including cirrhosis, HCC, and elevated all-cause mortality. When PYK2 is activated, it triggers dimerization associated with its N-terminal FERM domain, resulting in JNK signaling pathway activation. This process increases procollagen C-endopeptidase enhancer 1 in brown adipose tissue, thereby promoting liver fibrosis (79, 80). On the other hand, autophosphorylation of PYK2 at Tyr402 facilitates recognition by Src kinase, forming a PYK2-Src-RhoA ternary complex. This complex leads to YAP/TAZ activation and subsequent induction of connective tissue growth factor, which can also contribute to liver fibrosis (81). In conclusion, PYK2’s complex mechanisms in liver diseases shed light on the pathophysiological processes of hepatic disorders and suggest potential therapeutic targets for developing novel treatment strategies. Further research into PYK2’s molecular interactions and signaling pathways may yield significant advancements in liver disease management.
Acute pancreatitis
3.2.3
AP, a prevalent disorder of the digestive system, is characterized by complex and diverse pathogenesis. Research indicates that inflammatory responses, acinar cell damage, and inappropriate activation of trypsinogen constitute critical biological processes in AP (82). Under normal physiological conditions, pancreatic acinar cells synthesize and secrete digestive enzymes, with trypsinogen and other digestive enzymes remaining in an inactive state. However, when certain pathogenic factors disrupt this balance, leading to inappropriate activation of trypsinogen, acinar cell damage occurs, triggering inflammatory responses. In severe acute pancreatitis (SAP), reactive oxygen species (ROS) and oxidative stress have been closely associated with pancreatic acinar cell injury (83). During this process, PYK2 upregulates the expression of NADPH oxidases (NOXs), promoting excessive ROS generation. As highly reactive molecules, ROS can damage various intracellular biomolecules, including proteins, lipids, and nucleic acids, ultimately compromising cellular function. Furthermore, PYK2 facilitates the activation of AKT and MAPKs, exacerbating cellular oxidative stress and creating a more detrimental cellular environment (84–86). Additionally, PYK2 promotes increased expression of pro-inflammatory cytokines in macrophages (87). The substantial release of these pro-inflammatory cytokines exacerbates inflammatory damage in pancreatic tissue, manifesting as local tissue swelling, pain, and other inflammatory responses, thereby worsening the clinical condition.
PYK2 in metastasis and genomic alterations
4
PYK2 orchestrates metastasis in digestive cancers through multifaceted regulation of EMT, cell motility, and tumor microenvironment (TME) remodeling. In HCC, PYK2 promotes EMT by modulating Rac1/RhoA activity, enhancing invasive potential and distant dissemination (60). Concurrently, PYK2-driven PI3K/AKT activation upregulates VEGF, facilitating peritumoral angiogenesis to fuel metastatic spread (59). PDAC studies reveal that nuclear PYK2 scaffolds YAP/TAZ transcriptional complexes, sustaining cancer stemness and EMT via SOX9/OCT4 activation (18, 25). Furthermore, collagen-rich PDAC stroma engages DDR1-PYK2 signaling to induce N-cadherin expression and EMT, linking desmoplasia to metastasis (63, 64). Within the TME, PYK2 phosphorylates IRF5 in macrophages, amplifying pro-inflammatory cytokine release (e.g., IL-17A) and recruiting pathogenic CD11c^+^ macrophages in colitis-associated cancer (13, 71). Genomically, PTK2B (encoding PYK2) exhibits frequent amplifications and missense mutations in gastrointestinal malignancies (e.g., HCC, PDAC), correlating with metastatic progression and therapy resistance. For instance, hypoxia-induced HIF-1α transactivates TMEM237, which stabilizes NPHP1-PYK2 complexes to drive ERK1/2-dependent HCC metastasis (57). PYK2 also confers cisplatin resistance via PI3K/AKT-mediated anti-apoptotic signaling or mitochondrial Lon protein-triggered PYK2-SRC-STAT3 survival pathways (88, 89). Targeting PYK2 with inhibitors (e.g., PF-562,271) disrupts stromal crosstalk and reverses therapy resistance, highlighting its dual role as a metastatic scaffold and genomic driver.
Therapeutic targeting and overcoming resistance
5
PYK2 as a therapeutic target
5.1
PYK2 has been proven to exert essential functions in gastrointestinal cancers, serving as a signaling hub that integrates inputs from growth factors, integrins, and the tumor microenvironment to drive malignancy. Given its extensive physiological and pathological functions, close association with tumor development, potential roles in other diseases, and the ongoing clinical research on related therapeutic agents, PYK2 indeed represents a promising drug target. This multifaceted involvement positions PYK2 inhibition as a rational strategy for cancer therapy.
Classes of PYK2 inhibitors
5.2
Current PYK2-targeted therapeutics fall into three main categories (90): (1) ATP-competitive kinase inhibitors (e.g., PF-562,271, PF-431396), which bind the kinase domain and block phosphorylation (91); (2) allosteric inhibitors (e.g., T6BP), which disrupt scaffolding functions by targeting FERM domain dimerization (79); (3) PROTACs (Proteolysis-Targeting Chimeras), which are experimental agents (e.g., PYK2-PROTAC) that induce ubiquitin-mediated degradation (92). Ongoing clinical trials focus on PF-562,271 in pancreatic cancer (NCT04472174), while PROTACs remain preclinical (93).
Mechanisms of action of key inhibitors
5.3
PF-562,271 (Methane sulfonamide diaminopyrimidine), an ATP-competitive inhibitor (94), exhibits potent and reversible inhibition of PYK2 catalytic activity. By blocking PYK2 phosphorylation, it disrupts cancer-associated fibroblast persistence and monocyte recruitment, suppressing tumor growth and metastasis, suppressing tumor growth and metastasis (95). Combining this drug with sunitinib enhances PYK2 inhibition, yielding superior anti-angiogenic and anti-invasive effects (96). PF-431396 (trifluoromethyl pyrimidine) induces apoptosis, inhibits cell cycle progression and metastasis in PDAC and mesothelioma via PYK2 inhibition, and attenuates tumorigenicity in anchorage-independent conditions (97). It also blocks replication of adherent-invasive E. coli(AIEC) in Crohn’s disease and alters Salmonella infection course (98). T6BP blocks PYK2 FERM domain dimerization, attenuating JNK signaling. This reduces hepatic lipid accumulation and cytokine secretion, ameliorating fatty liver disease and fibrosis. T6BP also potentiates CBL-mediated PYK2 ubiquitination and degradation (79).
PYK2-mediated resistance mechanisms
5.4
Despite the growing body of research on PYK2 inhibitors, the potential development of acquired drug resistance poses new therapeutic challenges. Studies have shown that PYK2 overexpression increases cisplatin resistance, potentially through PI3K/AKT pathway activation, reduced apoptosis, and upregulation of drug resistance genes (89). Alternatively, cisplatin-induced mitochondrial DNA damage may lead to Lon (a stress protein) overexpression, which, upon binding with the Na+/Ca2+ exchanger, triggers mitochondrial calcium release into the cytoplasm, activating the PYK2-SRC-STAT3 pathway and subsequent BCL-2 expression, ultimately inhibiting apoptosis and contributing to cisplatin resistance (88). Regardless of the dominant mechanism, PYK2 inhibition can enhance tumor necrosis/apoptosis during cisplatin treatment, potentially reducing drug resistance.
Strategies to overcome resistance
5.5
Critically, PYK2 functions as a key mediator of acquired resistance to multiple chemotherapeutics, particularly cisplatin, in digestive tumors. Experimental evidence implicates PYK2 overexpression in conferring cisplatin resistance, primarily through two interconnected mechanisms: (1) Activation of the PI3K/AKT survival pathway, suppression of apoptosis, and upregulation of drug resistance genes (89); and/or (2) Cisplatin-induced mitochondrial stress leading to Lon protease overexpression, which triggers calcium release and subsequent activation of the PYK2-SRC-STAT3-BCL-2 anti-apoptotic axis (88). Consequently, targeted inhibition of PYK2 emerges as a rational strategy to overcome or prevent this acquired resistance, potentially sensitizing tumors to cisplatin and improving therapeutic outcomes, as suggested by studies combining PYK2 inhibitors with chemotherapy (95). Furthermore, given its role as a signaling hub and scaffold protein integrating inputs from growth factors, integrins, and the tumor microenvironment, PYK2 may also contribute to resistance against other therapeutic modalities, although this warrants further investigation in digestive cancer contexts. Therefore, targeting PYK2 is highly relevant for overcoming resistance to approved digestive cancer therapies. As a central node in key resistance/survival pathways (e.g., PI3K/AKT, STAT3) (34, 88, 89) and a scaffold for pro-tumorigenic signaling complexes (18, 34), PYK2 inhibition directly disrupts therapy evasion mechanisms. Preclinically, PYK2 inhibitors (e.g., PF-562,271, PF-431396) resensitize tumors to chemotherapeutics like cisplatin by counteracting anti-apoptotic signaling and compensatory survival cascades (95, 96). Furthermore, PYK2’s role in critical tumor microenvironment processes, including stromal crosstalk (e.g., DDR1-PYK2 in PDAC (63), macrophage polarization (e.g., IRF5 phosphorylation 13), and cancer stemness maintenance (e.g., via nuclear YAP/TAZ 18), suggests its contribution to resistance against broader therapies, potentially including targeted agents and immunotherapies. Strategies to mitigate FAK compensation, such as dual FAK/PYK2 inhibitors or exploiting PYK2-specific activation (Ca²^+^/PKC, oxidative stress, nuclear functions) (18, 27, 29, 34, 91, 95), enhance this approach’s feasibility. Thus, PYK2 targeting represents both a direct anti-tumor strategy and a promising combinatorial approach to restore efficacy of approved therapies facing acquired resistance in digestive cancers. While cisplatin’s cytotoxicity involves complex mechanisms beyond these two pathways (99), targeting PYK2 to circumvent drug resistance represents a promising avenue for future research.
Furthermore, given PYK2’s pivotal role as a signaling hub orchestrating multiple oncogenic pathways—including MAPK, PI3K-AKT, and YAP-TAZ—its inhibition presents a compelling rationale for combination therapies with agents targeting these downstream effectors, particularly for overcoming therapy resistance in digestive cancers. Preclinical evidence supports this approach: co-targeting PYK2 and VEGFR (sunitinib) demonstrated superior anti-angiogenic and anti-invasive effects compared to monotherapy in a hepatocellular carcinoma xenograft model, suggesting synergy in disrupting PYK2-driven tumor microenvironment remodeling and survival signaling (96). Specifically, combining PYK2 inhibitors with agents blocking the PI3K-AKT axis could counteract PYK2-mediated anti-apoptotic signaling and drug resistance gene upregulation observed in cisplatin resistance (88, 89). Equally promising is the combination with YAP-TAZ-TEAD pathway inhibitors, as nuclear PYK2 scaffolds transcriptional complexes (e.g., PYK2-Src-YAP/TAZ) critical for cancer stemness maintenance and EMT in PDAC and other GI malignancies (18, 25, 34). Targeting both the upstream activator (PYK2) and the key downstream transcriptional machinery (YAP-TAZ-TEAD) may offer a more comprehensive strategy to dismantle this resilience network and reverse therapy resistance. Future research should prioritize evaluating these rational combinations in models of innate and acquired resistance across diverse digestive tumors. The significant sequence homology (46% identity) and functional redundancy between PYK2 and FAK raise concerns that FAK may compensate for PYK2 inhibition, potentially limiting therapeutic efficacy (7, 8). This compensatory capacity is evidenced by enhanced suppression of tumor progression in HCC models when both kinases are co-inhibited compared to single targeting (41, 95). To circumvent this challenge, strategic approaches focus on dual inhibition or exploiting functional divergence. Dual FAK/PYK2 inhibitors (e.g., PF-562,271 derivatives) simultaneously block compensatory signaling nodes within this kinase family (91, 95). Alternatively, leveraging PYK2-specific regulatory mechanisms—such as its unique activation by Ca²^+^/PKC and oxidative stress (27, 29), and non-redundant nuclear scaffolding functions (e.g., β-catenin Y654 phosphorylation driving Wnt in PDAC; YAP/TAZ complex assembly sustaining cancer stemness) (18, 34) (18, 35)—enables context-selective targeting. This approach minimizes FAK-driven escape while capitalizing on tissue-specific roles of PYK2, such as its tumor-suppressive function in GC versus oncogenic actions in PDAC/HCC (38, 62).
PYK2 genomic alterations in digestive cancers
6
Emerging evidence implicates PYK2 (PTK2B) genomic alterations as drivers of digestive carcinogenesis. Analysis of public cohorts (TCGA, cBioPortal) reveals frequent PYK2 amplifications and missense mutations across gastrointestinal malignancies, often correlating with metastatic progression and therapy resistance. The table below summarizes key alterations and their clinical implications (Table 1):
Conclusion
7
Over the past decade, PYK2 has garnered significant attention in the scientific community as a critical regulatory molecule in various cellular processes. In addition to serving as a robust prognostic biomarker for tumor evaluation, this molecule exerts regulatory control over numerous critical signaling cascades that drive the pathogenesis and clinical manifestation of gastrointestinal disorders. This multifaceted role endows PYK2 with unique value in molecular therapy, diagnosis, and prognostic evaluation. Consequently, research on PYK2-targeted drugs has proliferated, particularly in combination with chemotherapeutic agents, demonstrating significant effects in slowing tumor progression, improving prognosis, and even modulating chemotherapy drug resistance. These results highlight the essential role of PYK2 in the onset and progression of diseases affecting the digestive system. However, considering PYK2’s involvement in multiple pathways, several critical questions remain to be addressed. These include potential cross-talk between these pathways, as well as the challenge of balancing antitumor efficacy with the reduction of complications and drug resistance risks in targeted therapy development. Therefore, the widespread application of PYK2-targeted therapy in digestive system diseases remains debatable and requires extensive clinical validation. Furthermore, compared to diseases in other systems, the research landscape between gastrointestinal disorders and PYK2 remains largely unexplored, particularly in specific diseases such as GC and CCA, indicating substantial room for further investigation. In conclusion, while the application prospects of PYK2 in digestive system disease treatment appear promising, it is imperative to maintain a scientifically rigorous approach and continuously promote high-quality clinical research to establish a solid foundation for its future clinical applications. Developing PYK2-targeted therapies requires in-depth research into their molecular mechanisms, possible side effects, and long-term effectiveness to ensure successful and safe clinical implementation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Shen T Guo Q. Role of pyk 2 in human cancers. Med Sci Monitor. (2018) 24:8172–82. doi: 10.12659/msm.913479, PMID: 30425234 PMC 6247758 · doi ↗ · pubmed ↗
- 2Wang J Bao P Liu Y. Pyk 2 regulates sepsis-induced lung injury via ferroptosis. Iran J Basic Med Sci. (2023) 26:1283–90. doi: 10.22038/ijbms.2023.69578.15153, PMID: 37886006 PMC 10598808 · doi ↗ · pubmed ↗
- 3Zheng J Suo L Zhou Y Jia L Li J Kuang Y. Pyk 2 suppresses contextual fear memory in an autophosphorylation-independent manner. J Mol Cell Biol. (2021) 13:808–21. doi: 10.1093/jmcb/mjab 057, PMID: 34529077 PMC 8782590 · doi ↗ · pubmed ↗
- 4Sbrana FV Fiordi B Bordini J Belloni D Barbaglio F Russo L. PYK 2 is overexpressed in chronic lymphocytic leukaemia: A potential new therapeutic target. J Cell Mol Med. (2023) 27:576–86. doi: 10.1111/jcmm.17688, PMID: 36747338 PMC 9930416 · doi ↗ · pubmed ↗
- 5López-Molina L Fernández-Irigoyen J Cifuentes-Díaz C Alberch J Girault J-A Santamaría E. Pyk 2 regulates MA Ms and mitochondrial dynamics in hippocampal neurons. Cells. (2022) 11:842–60. doi: 10.3390/cells 11050842, PMID: 35269464 PMC 8909471 · doi ↗ · pubmed ↗
- 6Momi S Canino J Vismara M Galgano L Falcinelli E Guglielmini G. Proline-rich tyrosine kinase Pyk 2 regulates deep vein thrombosis. Haematologica. (2022) 107:1374–83. doi: 10.3324/haematol.2021.279703, PMID: 35142150 PMC 9152972 · doi ↗ · pubmed ↗
- 7Zhu X Bao Y Guo Y Yang W. Proline-rich protein tyrosine kinase 2 in inflammation and cancer. Cancers (Basel). (2018) 10:139–52. doi: 10.3390/cancers 10050139, PMID: 29738483 PMC 5977112 · doi ↗ · pubmed ↗
- 8Shen T Guo Q. EGFR signaling pathway occupies an important position in cancer-related downstream signaling pathways of Pyk 2. Cell Biol Int. (2020) 44:2–13. doi: 10.1002/cbin.11209, PMID: 31368612 PMC 6973235 · doi ↗ · pubmed ↗