ABCA3 Surfactant‐Related Gene Variant Associated Interstitial Lung Disease in Adults: A Case Series and Review of the Literature
James Nolan, Jonathan Rodgers, John A. Mackintosh

TL;DR
This paper reports three adult cases of lung disease caused by ABCA3 gene variants, emphasizing the importance of genetic testing for unusual lung conditions.
Contribution
The study contributes three unique adult cases of ILD linked to compound heterozygous ABCA3 variants, expanding understanding of this rare condition in adults.
Findings
Three adult cases of ILD were found to be caused by compound heterozygous ABCA3 gene variants.
ABCA3-related lung disease is rare in adults but should be considered in cases of unusual ILD.
Early genetic testing is recommended for young adults with atypical interstitial lung disease.
Abstract
Surfactant‐related gene (SRG) variants are a rare but increasingly recognised cause of interstitial lung disease (ILD) in adults. Lung disease due to pathogenic variants in the adenosine triphosphate (ATP) binding cassette subfamily A member 3 (ABCA3) gene has been extensively described among infants and children but is rarely described in an adult population. The rarity and heterogeneity of lung disease due to ABCA3 variants raise significant challenges in recognition, diagnosis and management. In this case series we present three unique adult cases of ILD secondary to compound heterozygous ABCA3 variants, review the literature to provide an overview of this disease in an adult population and highlight the role for early genetic testing in young adults presenting with unusual ILD. Surfactant‐related gene variants are a rare but increasingly recognised cause of interstitial lung…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
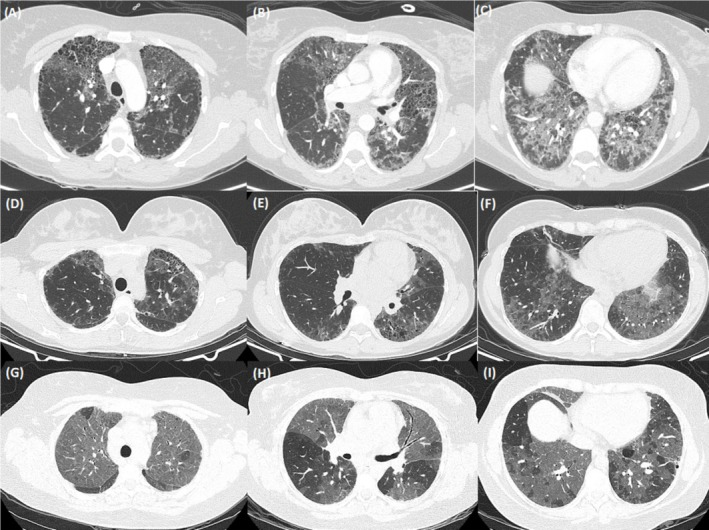 FIGURE 1
FIGURE 1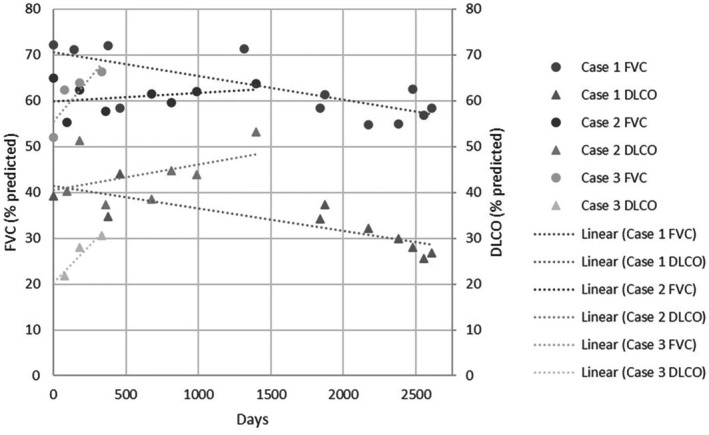 FIGURE 2
FIGURE 2| Participant | Age at presentation | Sex | Variant (c.DNA) | Variant (p.protein) | Literature |
|---|---|---|---|---|---|
| 1 | 39 | F | c.875A>T | p.(Glu292Val) | This study |
| c.4747C>T | p.(Arg1583Trp) | ||||
| 2 | 39 | F | c.875A>T | p.(Glu292Val) | |
| c.3997_3998del | p.(Arg1333Glyfs*24) | ||||
| 3 | 30 | F | c.875A>T | p.(Glu292Val) | |
| c.2063 T>C | p.(Leu688Pro) | ||||
| 4 | 52 | M | c.2891G>A | p.(Gly964Asp) | Campo [ |
| c.2891G>A | p.(Gly964Asp) | ||||
| 5 | 52 | M | c.2891G>A | p.(Gly964Asp) | |
| c.2891G>A | p.(Gly964Asp) | ||||
| 6 | 33 | F | c.2125C>T | p.(Arg709Trp) | Kröner [ |
| c.3579C>G | p.(Ile1193Met) | ||||
| 7 | 35 | F | c.[2921‐2922delinsCG;3079G>C] | p.[(Gly974Asp; Ala1027Pro)] | Legendre [ |
| c.[2921‐2922delinsCG;3079G>C] | p.[(Gly974Asp; Ala1027Pro)] | ||||
| 8 | 41 | M | c.875A>T | p.Glu292Val | Epaud [ |
| c.3081_3092delinsCG | p.Ser1028Valfs*103 | ||||
| 9 | 19 | F | c.838C>T | p.(Arg280Cys) | Klay [ |
| c.875A>T | p.(Glu292Val) | ||||
| 10 | 61 | F | c.875A>T | p.(Glu292Val) | |
| c.4451G>C | p.(Arg1484Pro) | ||||
| 11 | 77 | M | c.1675G>A | p.(Gly559Arg) | |
| c.4745C>G | p.(Thr1582Ser) | ||||
| 12 | 18 | F | c.737C>T | p.(Pro199Leu) | Tsuchiya [ |
| c.596C>T | p.(Pro246Leu) | ||||
| 13 | 18 | F | c.737C>T | p.(Pro199Leu) | |
| c.596C>T | p.(Pro246Leu) | ||||
| 14 | 43 | F | c.4873G>T | p.(Glu1625*) | Diesler [ |
| c.875A>T | p.(Glu292Val) | ||||
| 15 | 41 | F | c.58C>T | p.(Arg20Trp) | |
| c.4706_4708del | p.(lle1569del) | ||||
| 16 | 30 | F | c.4237G>A | p.(Gly1413Ser) | |
| c.4444C>T | p.(Arg1482Trp) | ||||
| 17 | 19 | F | c.2068G>A | p.(Glu690Lys) | |
| c.875A>T | p.(Glu292Val) | ||||
| 18 | 50 | F | c.838C>T | p.(Arg280Cys) | |
| c.4984‐2A>C | p.? | ||||
| 19 | 42 | F | c2414 + 1G>C | p.? | |
| c.875A>T | p.(Glu292Val) | ||||
| 20 | 38 | M | c.3081_3092delinsCG | p.(Ser1028Valfs*103) | |
| c.875A>T | p.(Glu292Val) | ||||
| 21 | 19 | M | c.4483‐4507del | p.(Val1495Cysfs*21) | |
| c.875A>T | p.(Glu292Val) | ||||
| 22 | 35 | F | c.127C>T | p.(Arg43Cys) | |
| c.3004G>A | p.(Gly1002Ser) | ||||
| 23 | 19 | F | c.347T>C | p.(Phe116Ser) | |
| c.838C>T | p.(Arg280Cys) |
| DNA position | Protein | Number of Subjects with Mutation | Variant classification | Mutation Type | Exon | ClinVar reference (if present) | Reference, if not on ClinVar |
|---|---|---|---|---|---|---|---|
| c.58C>T | Arg20Trp | 1 | Likely pathogenic | Missense | 5 | — | Diesler [ |
| c.127C>T | Arg43Cys | 1 | Likely pathogenic | Missense | 5 | VCV001767530 | |
| c.347 T>C | Phe116Ser | 1 | Likely pathogenic | Missense | 6 | — | Diesler [ |
| c.596C>T | Pro199Leu | 2 | Likely pathogenic | Missense | 7 | VCV002982329 | |
| c.737C>T | Pro246Leu | 2 | Likely pathogenic | Missense | 8 | VCV002982329 | |
| c.838C>T | Arg280Cys | 3 | Pathogenic | Missense | 8 | VCV000318566 | |
| c.875A>T | Glu292Val | 11 | Pathogenic | Missense | 9 | VCV000203381 | |
| c.1675G>A | Gly559Arg | 1 | Variant of uncertain significance (VUS) | Missense | 14 | VCV003341964 | |
| c.2063 T>C | Leu688Pro | 1 | Pathogenic | Missense | 17 | — | Diesler [ |
| c.2068G>A | Glu690Lys | 1 | VUS | Missense | 17 | VCV003629580 | |
| c.2125C>T | Arg709Trp | 1 | Conflicting classifications of pathogenicity | Missense | 17 | VCV000162674 | |
| c.2414 + 1G>C | ? | 1 | Likely pathogenic | Splice site | Intron | — | Diesler [ |
| c.2891G>A | Gly964Asp | 2 | Likely pathogenic | Missense | 21 | — | Campo [ |
| c.2921‐2922delinsCG | Gly974Asp | 1 | VUS | Missense | 21 | — | Legendre [ |
| c.3004G>A | Gly1002Ser | 1 | Likely pathogenic | Missense | 21 | — | Diesler [ |
| c.3079G>C | Ala1027Pro | 1 | VUS | Missense | 22 | — | Legendre [ |
| c.3081_3092delinsCG | Ser1028Valfs*103 | 2 | Likely pathogenic | Frameshift variant | 22 | — | Diesler [ |
| c.3579C>G | Ile1193Met | 1 | VUS | Missense | 24 | VCV002137767 | |
| c.3997_3998del | Arg1333Glyfs*24 | 1 | Pathogenic | Frameshift variant | 26 | VCV001317554 | |
| c.4237G>A | Gly1413Ser | 1 | Likely pathogenic | Missense | 28 | — | Diesler [ |
| c.4444C>T | Arg1482Trp | 1 | Conflicting classifications (VUS in ClinVar, Pathogenic in Diesler) | Missense | 29 | VCV003375130 | Diesler [ |
| c.4451G>C | Arg1484Pro | 1 | VUS | Missense | 29 | — | Klay [ |
| c.4483‐4507del | Val1495Cysfs*21 | 1 | Pathogenic | Frameshift variant | 29 | VCV002631175 | |
| c.4706_4708del | lle1569del | 1 | Pathogenic | In frame deletion | 30 | — | Diesler [ |
| c.4745C>G | Thr1582Ser | 1 | VUS | Missense | 31 | VCV003341823 | |
| c.4747C>T | Arg1583Trp | 1 | Likely pathogenic | Missense | 31 | — | Our study |
| c.4873G>T | Glu1625* | 1 | Likely pathogenic | Stop gain | 31 | — | Diesler [ |
| c.4984‐2A>C | ? | 1 | Likely pathogenic | Splice variant | Intron | — | Diesler [ |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeonatal Respiratory Health Research · Medical Imaging and Pathology Studies · Interstitial Lung Diseases and Idiopathic Pulmonary Fibrosis
Introduction
1
Surfactant‐related gene (SRG) variants predominantly cause respiratory disease in the paediatric population but are increasingly recognised as a rare cause of interstitial lung disease (ILD) in adults [1, 2]. Infants with biallelic loss of function variants in the adenosine triphosphate (ATP) binding cassette subfamily A member 3 (ABCA3) gene present with neonatal respiratory failure and death by 1 year of age [1, 3]. Other combinations of biallelic ABCA3 variants (e.g., missense, splicing, or in‐frame insertion/deletions) resulting in adult ILD have been reported in case reports and small cohort studies, but clinical information is limited [4, 5]. Herein, we describe three unique adult cases of ILD secondary to ABCA3 variants and review the literature to provide clinical and radiological clues to establishing a diagnosis and discuss management.
Case Series
2
Case 1
2.1
A 39‐year‐old female was admitted to hospital with urosepsis, at which time she was noted to have clubbing, with incidental fibrotic lung changes on abdominal computer tomography (CT) imaging. She had a medical history of hypertension, depression, and chronic back pain, with a nominal diagnosis of asthma and an active smoking history of 25 pack‐years. Dedicated pulmonary imaging showed extensive honeycombing cystic formation in a non‐specific distribution (Figure 1). Spirometry was moderately restrictive with FVC 2.75 L (72% predicted) and severely reduced gas diffusion with DLCO 39% predicted (Figure 2). A wedge biopsy was undertaken with histology favouring fibrosing non‐specific interstitial pneumonia (NSIP) with possible early usual interstitial pneumonia. During a subsequent hospitalisation with acute hypoxic respiratory failure, interval CT demonstrated the development of extensive ground glass opacification (GGO) and treatment with azathioprine and oral steroids was initiated. Over the following 4 years, the patient suffered frequent hospitalisations with acute and infective exacerbations of ILD. Maintenance immunosuppression was changed to mycophenolate, pulmonary function continued to decline and domiciliary oxygen was supplied for progressive hypoxia. Lung transplantation assessment was prevented by steroid‐associated obesity and intermittent smoking. Six years after initial ILD diagnosis, the patient was hospitalised with acute decompensated pulmonary hypertension. Two years later, genetic testing of a panel of genes associated with ILD identified one pathogenic and one likely pathogenic variant in ABCA3 (c.875A>T, p.(Glu292Val) and c.4747C>T, p.(Arg1583Trp); NM_001089.3 respectively) [3]. An additional variant of uncertain significance (VUS), ABCA3 c.235G>A, p.(Glu79Lys), was also identified in the gene, which is predicted to be deleterious by most in silico tools. Parental samples were not available for phasing variants. In the context of progressive pulmonary decline, lung transplantation was revisited, but during assessment, the patient was diagnosed with extensive stage small cell lung cancer. Due to deteriorating performance status, best supportive care was provided and she died 3 months later, 9 years after initial identification of ILD.
Case series of three females with ILD secondary to ABCA3 variants. Representative CT slices at presentation from (A–C) Case 1, (D–F) Case 2, and (G–I) Case 3.
Lung function trajectory from first presentation.
Case 2
2.2
A 39‐year‐old female was referred to the outpatient clinic with 10 years of progressive dyspnoea. She had a nominal diagnosis of asthma and was the mother of two children delivered without complication. There was no family history of pulmonary disease. On initial review, she was noted to be clubbed but with normal SpO_2_ on room air. Spirometry demonstrated moderate restriction with FVC 1.83 L (55% predicted) and severely reduced gas diffusion with DLCO 40% predicted (Figure 2). CT thorax demonstrated patchy GGO surrounding numerous thin‐walled cysts with some areas of co‐existent cylindrical destruction bordering on honeycombing (Figure 1). An ILD gene panel revealed a pathogenic frameshift variant (ABCA3:c.3997_3998del; p.(Arg1333Glyfs*24)) and a pathogenic missense variant (ABCA3:c.875A>T; p.(Glu292Val)). Parental testing confirmed that each parent was a carrier of one of the variants, and the patient was therefore compound heterozygous. Antifibrotic therapy, nintedanib, was initiated, but radiological progression was noted on serial imaging over 2 years. Hydroxychloroquine (HCQ) was subsequently prescribed, with 1‐year interval radiology showing reduction in GGOs, but progressive focal fibrotic changes. However, clinical progress, spirometry, and gas diffusion remained relatively stable over a further 3‐year period, and the patient continues to work full‐time in a clerical role.
Case 3
2.3
A 30‐year‐old female was referred to their local hospital service with 3 years of progressive dyspnoea. She was a never smoker, without significant past medical history and the mother of three children born without complication. There was no family history of pulmonary disease. Notable reported exposures were household mould and an Alexandrine parrot of 10 years. At the time of initial assessment, she was noted to be clubbed with resting SpO_2_ 91% on room air. Spirometry demonstrated moderate restriction with FVC 1.98 L (62% predicted) and severely reduced gas diffusion with DLCO 22% predicted (Figure 2). A 6‐min walk test (6MWT) showed significantly reduced walk distance at 330 m and marked exertional hypoxia with nadir SpO_2_ 74%. CT of the thorax revealed extensive GGO with mosaic attenuation, areas of minor subpleural cysts, prominent mediastinal and hilar lymphadenopathy, and dilatation of the pulmonary arteries (Figure 1). Autoimmune serology was non‐contributory. Bronchoscopy was performed with bronchoalveolar lavage cytology demonstrating 9% lymphocytes and 7.5% eosinophils; transbronchial lymph node aspirate showed no abnormality. She subsequently presented to a peripheral hospital 3 months later with progressive dyspnoea and hypoxic respiratory failure. Interval CT imaging was stable. Broad‐spectrum antibiotics were administered followed by methylprednisolone 1000 mg intravenous (IV) daily for 3 days, then oral prednisolone 50 mg daily without benefit. The patient was subsequently transferred to a tertiary centre. Echocardiogram demonstrated a mildly dilated right ventricle (RV) with preserved systolic function and mildly dilated main pulmonary arteries. Borderline elevated pulmonary pressures with a mean pulmonary artery pressure of 21 mmHg were found on right heart catheterisation. Genetic testing was requested, and treatment withHCQ 200 mg twice daily was initiated. Domiciliary oxygen was arranged. The results of the 40 gene ILD panel demonstrated a pathogenic missense variant (c.875A>T, p.(Glu292Val)) and a VUS (c.2063 T>C, p.(Leu688Pro)) in ABCA3. Parental samples were not available for phasing variants. While a VUS does not represent a genetically confirmed diagnosis, the available molecular evidence leant towards the variant being pathogenic and her presentation was consistent with an SRG‐related condition. Steroids were weaned to cessation; HCQ was continued and the patient was referred for lung transplantation assessment.
Discussion
3
Lung disease due to ABCA3 variants is inherited in an autosomal recessive fashion. Over 300 ABCA3 pathogenic variants have been identified. Compound heterozygosity is reported in more than three‐quarters of patients, with a familial history of ILD in approximately a quarter of cases [4, 6]. Pathogenic variants in ABCA3, which encodes a lipid transporter protein crucial for surfactant production in alveolar type II cells, result in disrupted surfactant homeostasis and progressive ILD [7]. Not all variants result in complete loss of function, and there is an increasing cellular‐based understanding of ABCA3 variants beyond that of intracellular protein mis‐trafficking and lipid transportation dysfunction [1, 2, 8]. Disease is predominantly associated with lethal neonatal respiratory distress and paediatric ILD, with 5‐year survival less than 20% [1, 9]. There are rare case reports of children surviving into adulthood, and across the literature, fewer than 50 reports of ABCA3‐associated ILD diagnosed in adults [1, 4, 10].
While adult diagnosed disease appears to be a heterogenous group, with highly variable disease characteristics, there are some emerging trends within the literature [5]. Onset of disease in SRG related adult ILD is difficult to ascertain due to disease rarity but appears to clinically occur at a younger age (median 45 years) than more recognised causes such as idiopathic pulmonary fibrosis (median 65 years). ABCA3 associated ILD presenting in an adult population appears to have an even earlier age of identification (median age 31.5 years), with a possible female predominance [4].
Almost all patients present with dyspnoea and over half with a non‐productive cough, 80% have crackles on initial pulmonary examination and clubbing is reported in two‐thirds. Over one‐quarter of reported cases were current or former smokers [4]. Respiratory function tests in ABCA3 pathogenic variant‐related ILD demonstrate a moderate restrictive pattern in almost all patients at the time of diagnosis, with a moderate to severe reduction in gas diffusion. These findings are generally more severe than in other SRG‐related ILD at presentation; however, progressive annual decline occurs at a lower rate [4].
Radiological findings are most commonly that of an unclassifiable ILD with GGO predominance. Pulmonary cysts are reported in over 90%, lymphadenopathy, reticulation and fibro‐elastosis features in over 80%, with honeycombing a rare feature [4, 5, 11, 12]. It has been hypothesised that imaging follows a progression from GGO to cystic disease and reticulations prior to the establishment of fibrosis [5]. Histopathology is predominantly NSIP, with the presence of mild alveolar epithelial type II cell hyperplasia and increased alveolar macrophages [4, 11].
Overall survival for patients with ABCA3‐related disease is unknown, but there appears to be a better prognosis and median survival than other SRG‐related ILD, which has been reported as 10 years from diagnosis. ILD exacerbations occur in just over one‐third of patients over 10 years, with lower rates of reported pulmonary hypertension than in other SRG disorders [4]. There is a recognised increased risk of lung cancer in individuals with SRG‐ILD and growing identification of the role of surfactant‐associated gene variants in individuals with lung cancer [13, 14, 15]. However, while lung cancer was identified in Case 1, noting a significant smoking history, there is no known association between ABCA3 pathogenic variant‐related ILD and lung cancer. Given the rarity of this disease and increasing identification of association with other surfactant‐associated gene variants, further consideration of a potential link is required.
Treatment for paediatric ABCA3‐related ILD traditionally involves immunomodulatory drugs, which have been shown to provide initial improvements in FVC in adult patients, but minimal sustained benefit [16]. HCQ has provided reported clinical improvement in case studies with some suggestion efficacy may be ABCA3 variant specific [16, 17, 18]. Treatment with cystic fibrosis transmembrane conductance regulator (CFTR) potentiators ivacaftor and genistein has been trialled, with suggestion it may rescue ABCA3 phospholipid transport protein functionality [19, 20].
Genetic screening considerations for all direct family members are recommended. It has been suggested that asymptomatic carriers of pathogenic variants should undergo yearly clinical screening and respiratory function tests with 5‐yearly chest radiology [5].
The pathogenic variant c.875A>T, p.(Glu292Val) was identified in all three cases presented herein. The allele frequency of this pathogenic variant is 0.00453 in gnomAD v4, seen 7316 times, with 20 homozygotes [1, 2]. This missense variant is a functional hypomorph resulting in moderately impaired ABCA3 function [1, 2, 21, 22]. Glu292Val appears to be the most commonly reported pathogenic ABCA3 variant among adults (Tables 1 and 2).
In this series, we present three unique cases of young adult females with ABCA3‐associated ILD, presenting with functional limitation, markedly deranged pulmonary physiology, and grossly abnormal CT imaging. There were significant delays to definitive diagnosis, with reported symptoms from 3 to 10 years and purported diagnoses of asthma. Empiric treatment did not provide clinical benefit, with significant immunosuppressive side effects, in the instance of Case 1, contributing to a delay in transplantation assessment. Early genetic testing, in young adults presenting with unusual ILD, may improve diagnosis, treatment, and outcomes for these patients.
In conclusion, the three presented cases demonstrate the variability of clinical presentation, investigative findings, and disease progression in adult patients with ABCA3‐related ILD. A more comprehensive understanding of ABCA3‐related ILD in adults is required. This series demonstrates the potential value for prompt upfront genetic testing in young people with unusual ILD, as opposed to empiric therapy.
Author Contributions
All authors contributed equally to the drafting of this manuscript and approve its content.
Consent
The authors declare that appropriate written informed consent was obtained for the publication of this manuscript and accompanying images.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1C. Kröner , T. Wittmann , S. Reu , et al., “Lung Disease Caused by ABCA 3 Mutations,” Thorax 72, no. 3 (2017): 213–220.27516224 10.1136/thoraxjnl-2016-208649 · doi ↗ · pubmed ↗
- 2M. A. Coghlan , A. Shifren , H. J. Huang , et al., “Sequencing of Idiopathic Pulmonary Fibrosis‐Related Genes Reveals Independent Single Gene Associations,” BMJ Open Respiratory Research 1, no. 1 (2014): e 000057.10.1136/bmjresp-2014-000057 PMC 426508325553246 · doi ↗ · pubmed ↗
- 3J. A. Wambach , A. M. Casey , M. P. Fishman , et al., “Genotype‐Phenotype Correlations for Infants and Children With ABCA 3 Deficiency,” American Journal of Respiratory and Critical Care Medicine 189, no. 12 (2014): 1538–1543.24871971 10.1164/rccm.201402-0342 OCPMC 4226019 · doi ↗ · pubmed ↗
- 4R. Diesler , M. Legendre , S. Si‐Mohamed , et al., “Similarities and Differences of Interstitial Lung Disease Associated With Pathogenic Variants in SFTPC and ABCA 3 in Adults,” Respirology 29, no. 4 (2024): 312–323.38345107 10.1111/resp.14667 · doi ↗ · pubmed ↗
- 5D. Klay , J. C. Grutters , J. J. van der Vis , et al., “Progressive Disease With Low Survival in Adult Patients With Pulmonary Fibrosis Carrying Surfactant‐Related Gene Mutations: An Observational Study,” Chest 163, no. 4 (2023): 870–880.36370864 10.1016/j.chest.2022.11.002 · doi ↗ · pubmed ↗
- 6M. F. Beers and S. Mulugeta , “The Biology of the ABCA 3 Lipid Transporter in Lung Health and Disease,” Cell and Tissue Research 367, no. 3 (2017): 481–493.28025703 10.1007/s 00441-016-2554-z PMC 5321817 · doi ↗ · pubmed ↗
- 7C. H. M. Van Moorsel , J. J. Vis , and J. C. Grutters , “Genetic Disorders of the Surfactant System: Focus on Adult Disease,” European Respiratory Review 30, no. 159 (2021): 200085.33597124 10.1183/16000617.0085-2020 PMC 9489129 · doi ↗ · pubmed ↗
- 8Y. L. Sun , E. E. Hennessey , H. Heins , et al., “Human Pluripotent Stem Cell Modeling of Alveolar Type 2 Cell Dysfunction Caused by ABCA 3 Mutations,” Journal of Clinical Investigation 134, no. 2 (2024): e 164274, 10.1172/JCI 164274.38226623 PMC 10786693 · doi ↗ · pubmed ↗