Generation and Nitric Oxide Reactivity of a Cobalt(II) Superoxide Complex via Guanidine-Based Ligand Non-Innocence
Dibya Jyoti Barman, Thomas Lohmiller, Konstantin Krause, Sagie Katz, Michael Haumann, Peter Hildebrandt, Kallol Ray

TL;DR
A new cobalt complex was created that activates oxygen and reacts with nitric oxide to nitrate its own ligand, thanks to a special guanidine-based ligand.
Contribution
The study introduces a novel cobalt(II) complex with a redox-active guanidine-based ligand that enables O2 activation and NO reactivity without changing the cobalt oxidation state.
Findings
A cobalt(II) complex forms a μ-hydroxo-bridged product via a superoxide intermediate without altering the cobalt oxidation state.
The same reaction occurs with a zinc analogue, highlighting the ligand's role in O2 activation.
The cobalt–superoxide intermediate reacts with nitric oxide to form a peroxynitrite intermediate that nitrates the ligand's phenyl groups.
Abstract
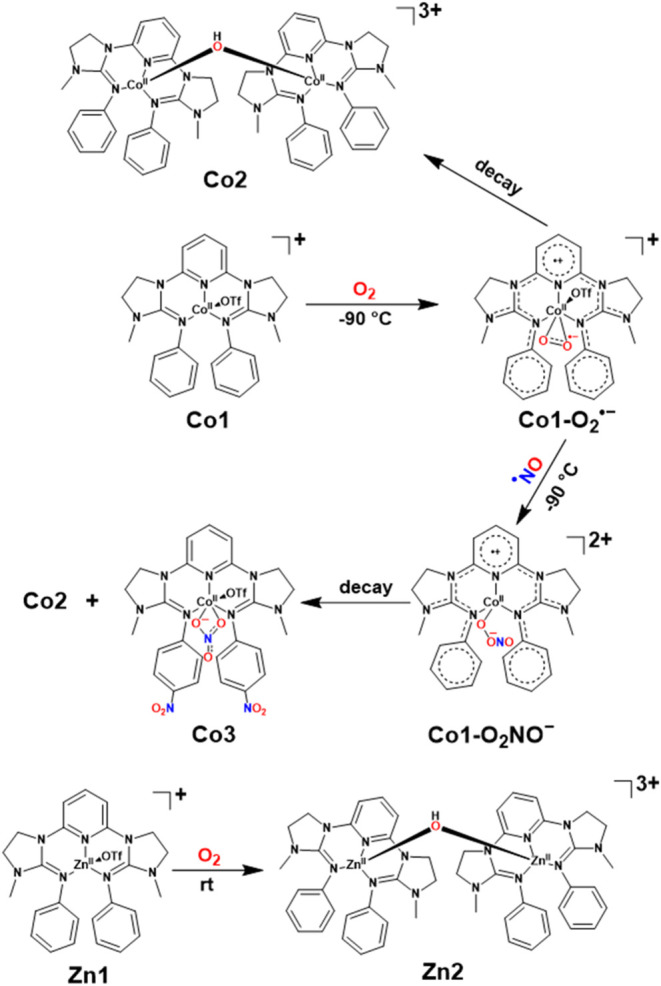
A novel guanidine-based NNN pincer cobalt(II) complex, Co1, was investigated for dioxygen (O2) activation. Upon reaction with O2 at room temperature, a μ-hydroxo-bridged CoII–OH-CoII complex (Co2) was isolated as the final oxygenated product. In-depth spectroscopic investigation revealed that the formation of Co2 occurs via an intermediate superoxide species (Co1–O 2 •– ), by taking advantage of the redox non-innocence behavior of the ligand (3,3′-(pyridine-2,6-diyl)bis(1-methyl-N-phenylimidazolidin-2-imine)), and therefore keeping the cobalt oxidation state unchanged at +2. Interestingly, the zinc analogue, Zn1, was also shown to yield a similar μ-hydroxo-bridged ZnII–OH-ZnII complex (Zn2) as the final product under the same aerobic conditions, providing further confirmation of the ligand influence on metal oxidation and spin states during O2 activation. Further reactivity…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1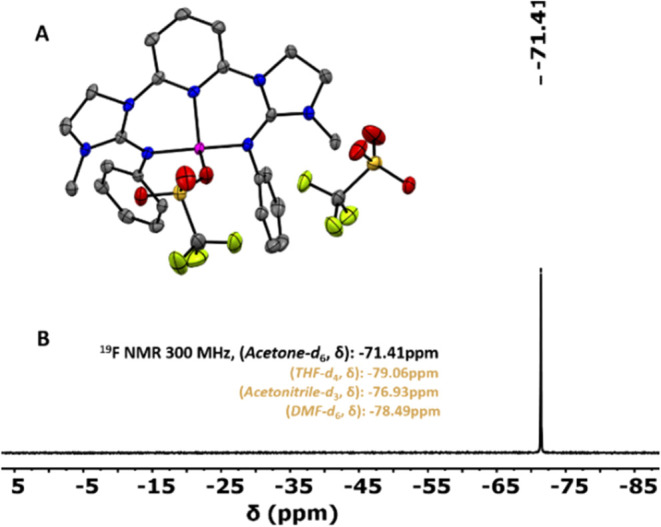 1
1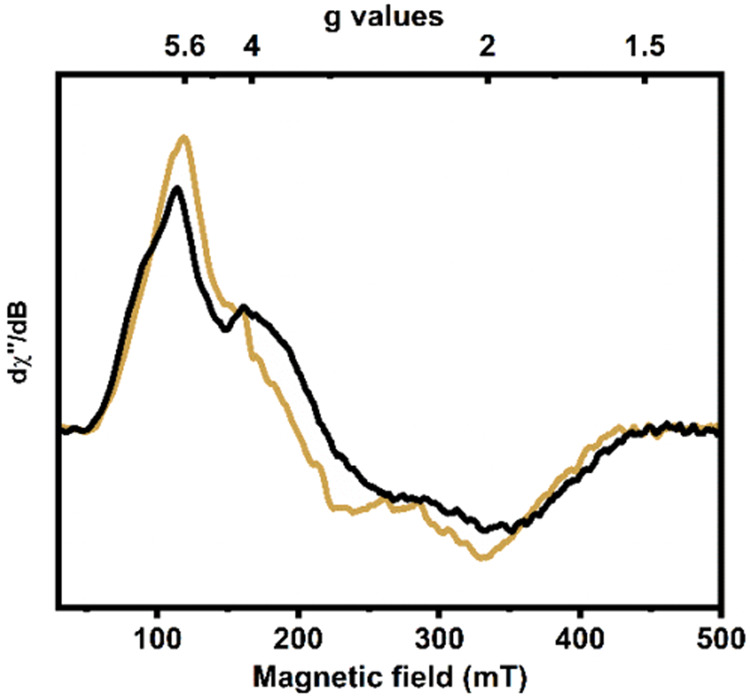 2
2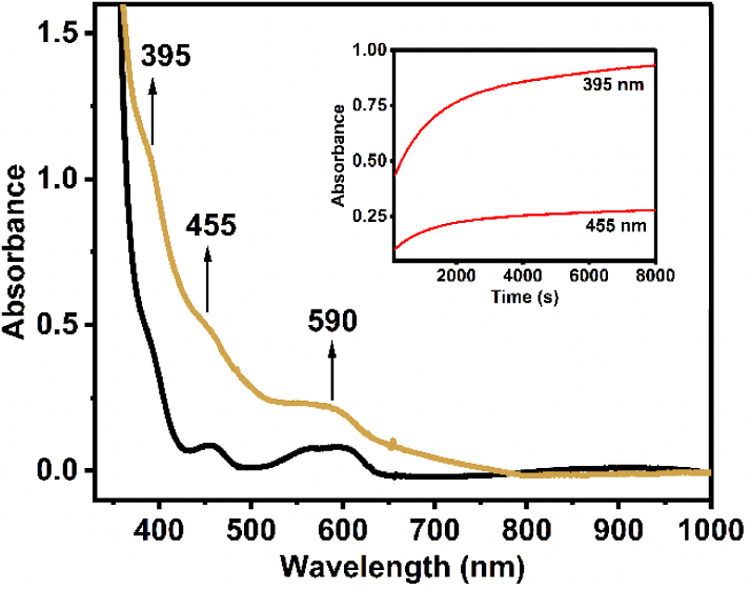 3
3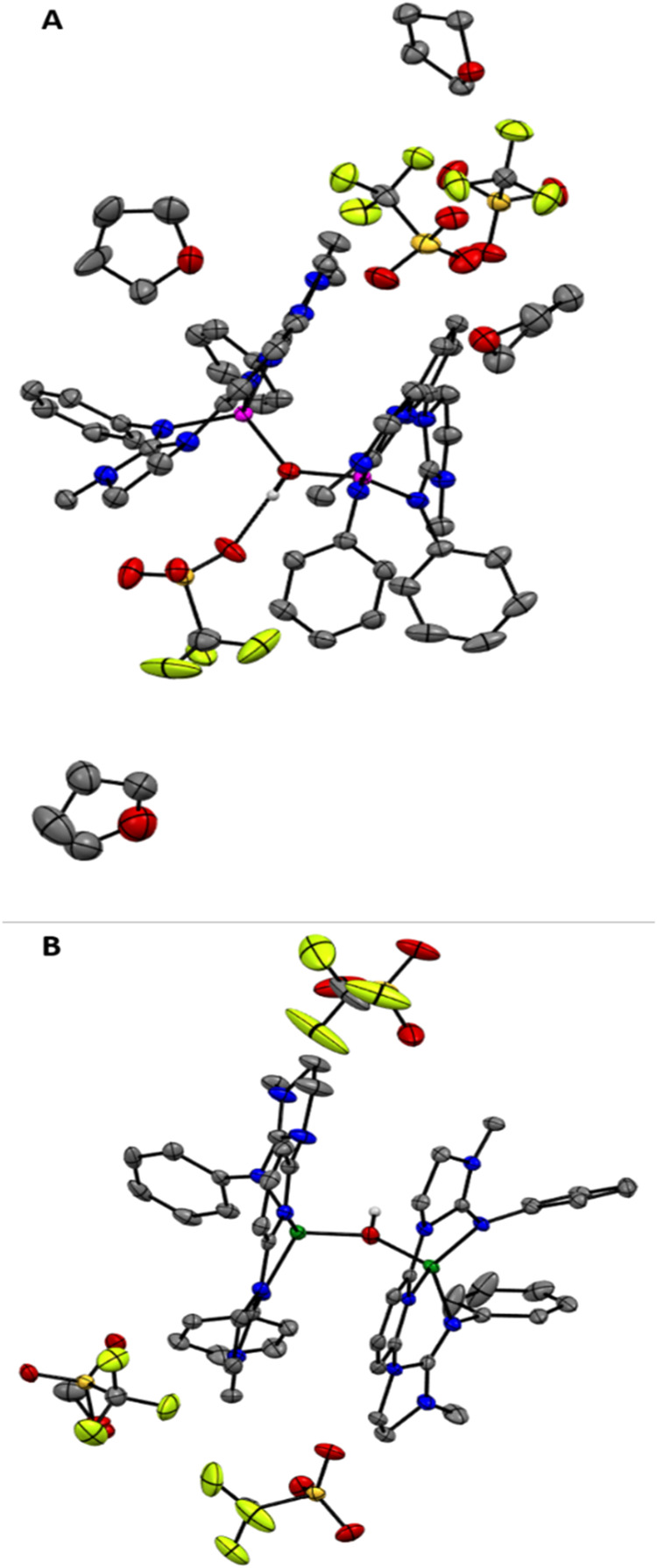 4
4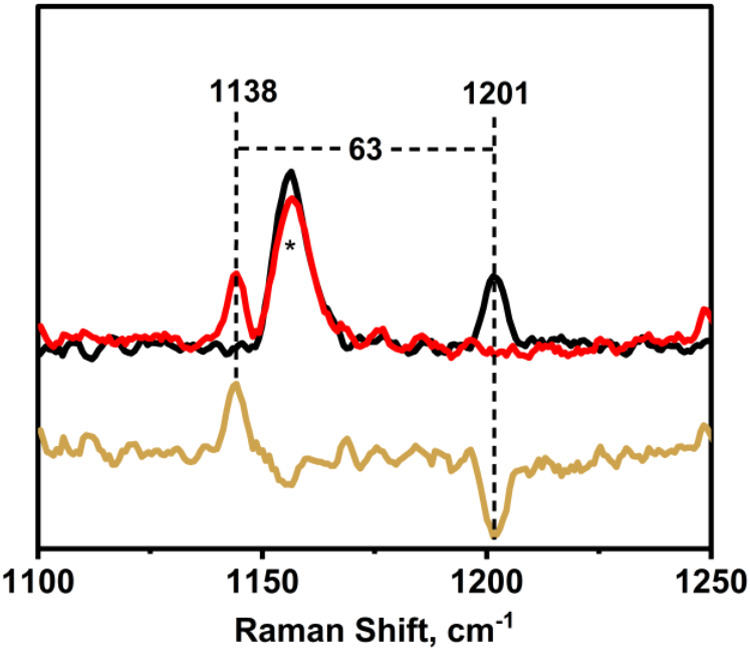 5
5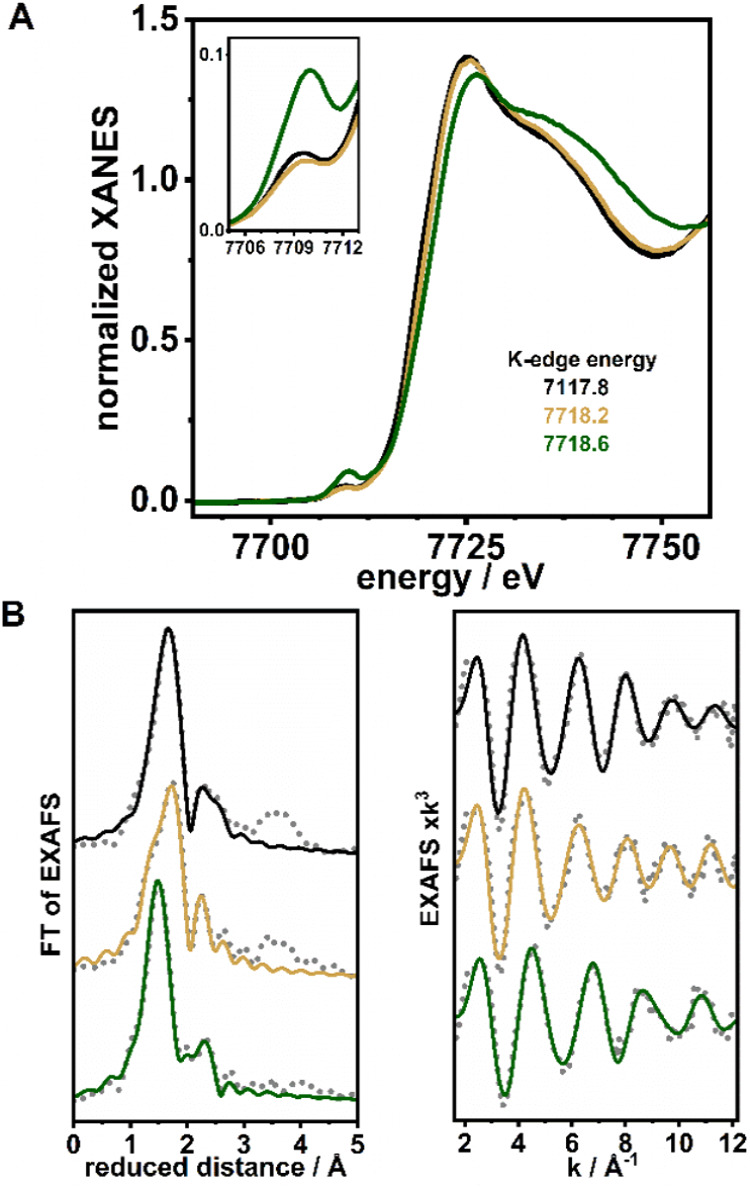 6
6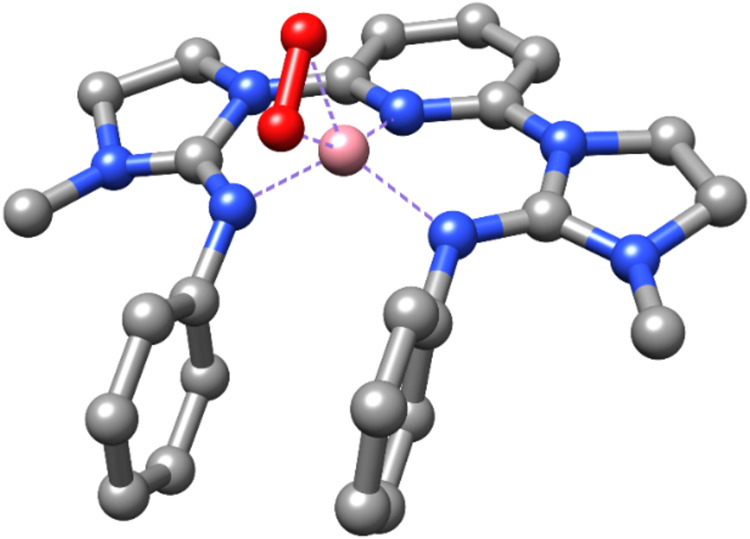 7
7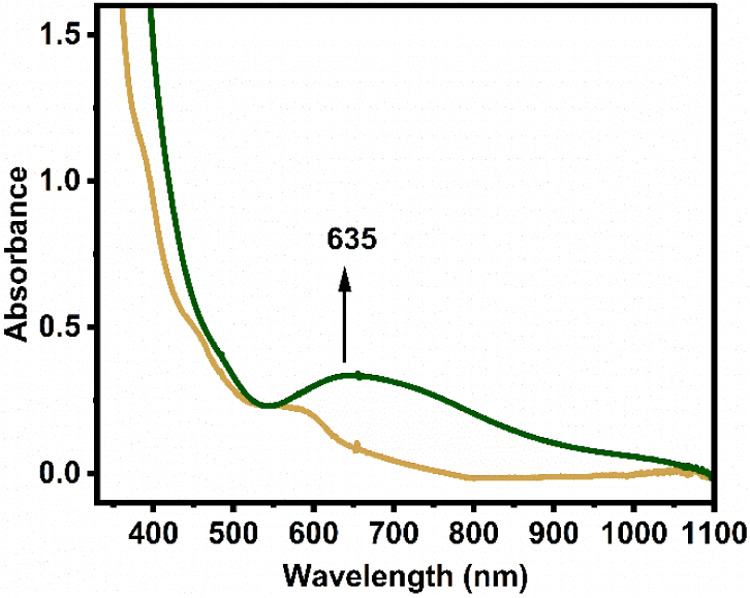 8
8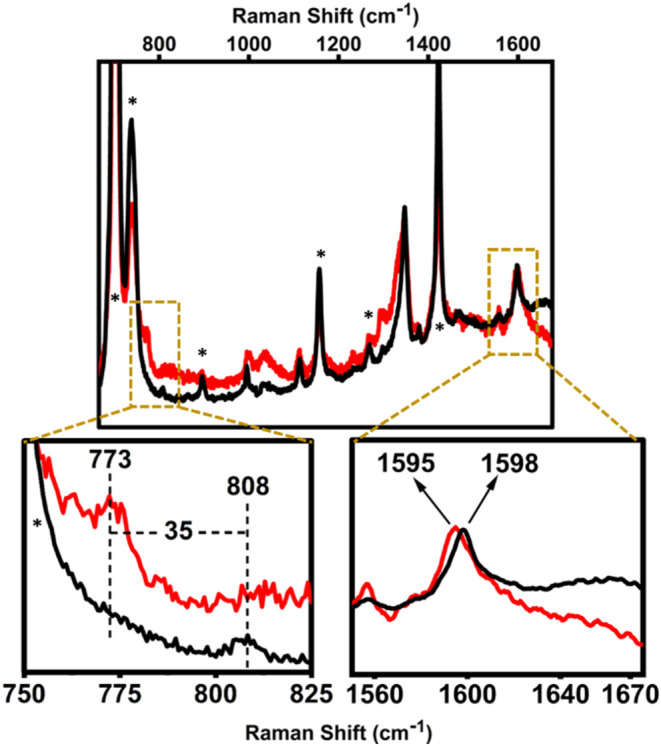 9
9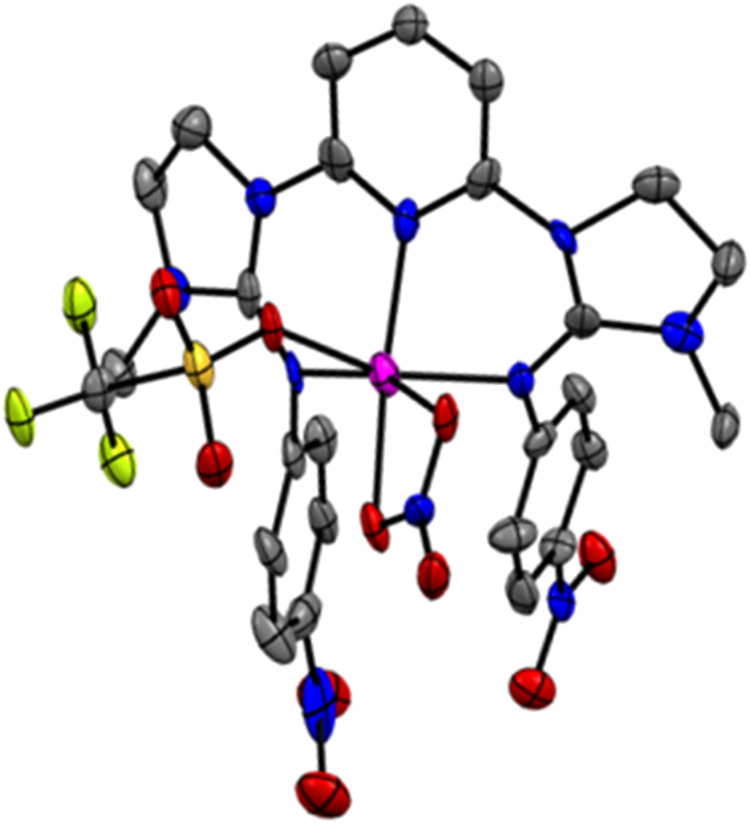 10
10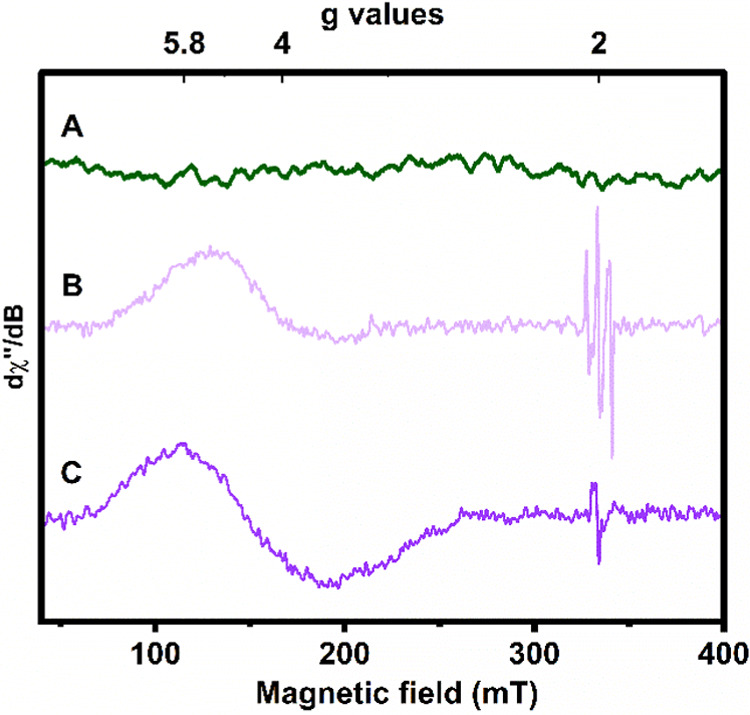 11
11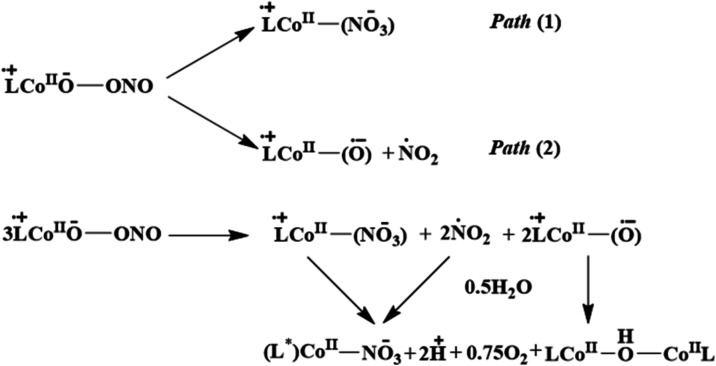 2
2- —Deutsche Forschungsgemeinschaft10.13039/501100001659
- —Deutsche Forschungsgemeinschaft10.13039/501100001659
- —Deutsche Forschungsgemeinschaft10.13039/501100001659
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsElectrochemical sensors and biosensors · Electrochemical Analysis and Applications · Oxidative Organic Chemistry Reactions
Introduction
Peroxynitrite (^ – ^OONO) is an incredibly reactive species that is generated by the near diffusion-controlled combination of nitric oxide (^•^NO) and the superoxide (O_2_ ^•–^) anion. ?,? It is a fascinating mediator of nitric oxide biochemistry and oxidative/nitrative stress injury. ?−? ? ? ? ? ? Metal ions in biological systems are key players in the generation and stabilization of OONO^–^, which undergo various thermal transformation reactions like isomerization to nitrate (NO_3_ ^–^) or activation toward substrate oxidation/nitration reactions. ?−? ? ? ? ? Heme proteins have been extensively investigated in terms of their role in peroxynitrite formation and the subsequent transformation to nitrate. ?,?,? For example, nitric oxide dioxygenases (NOD) are known to be important enzymes that convert ^•^NO to nitrate using O_2_, potentially via peroxynitrite intermediates. ?,? The biological relevance of ^ – ^OONO inspired a wide range of theoretical and experimental studies of its coordination and interaction with transition metal centers and subsequently, its reactivity. However, the formation of discrete metal-peroxynitrite complexes is a rare occurrence, but they have been suggested to form as transient intermediates from metal-NO + O_2_(g) or metal-O_2_ + ^•^NO(g) reactions. ?−? ? ? ? ? ? ? ?
Following our interest in cobalt oxidative chemistry, we note that the literature on solution chemistry of the cobalt ion with peroxynitrite is limited; no examples of isolable cobalt(II)-peroxynitrite species with the direct use of O_2_ and ^•^NO have been described. Clarkson and Basolo first reported the chemistry of a cobalt-nitrosyl complex with O_2_, in which a transient cobalt(III)-peroxynitrite intermediate was proposed to form the nitrite-bound product.? Similarly, Co(III)-nitrosyl complexes of 12-, 13-, and 14-membered N-tetramethylated cyclam ligands, [(12-TMC)Co^III^(NO)]^2+^, [(13-TMC)Co^III^(NO)]^2+^, and [(14-TMC)Co^III^(NO)]^2+^ were reported to react with the superoxide radical and O_2_, respectively, resulting in Co(II)-nitrite/nitrate, where the involvement of a Co(III)-peroxynitrite intermediate was presumed. ?,? In addition, Mondal and co-workers suggested the formation of a transient and nonisolable Co(II)-peroxynitrite intermediate in the reaction of a Co(III)-peroxo species with ^•^NO. ?,? The formation of cobalt-peroxynitrite chemistry is also biologically relevant; cobalamins (Cbls) are known to react with ^•^NO to yield nitrosocobalamins (NOCbls), which further react with O_2_ to form nitrocobalamin through the presumable formation of peroxynitrite Co(III) intermediate. ?−? ? The formation of cobalt-peroxynitrite is initiated in most cases upon addition of an O_2_ or ^•^NO at Co^II^ centers to yield kinetically inert low-spin terminal Co^III^ superoxide or nitrosyl moieties, which then react with ^•^NO or O_2_/O_2_ ^•–^/O_2_ ^2–^, respectively, to form Co-OONO moieties.
Herein, we report the synthesis of a novel cobalt(II) complex, Co1, supported by a guanidine-based NNN pincer ligand (L = 3,3′-(pyridine-2,6-diyl)bis(1-methyl-N-phenylimidazolidin-2-imine)), which enables the activation of O_2_ to superoxide via ligand-based electron transfer, leading to a cobalt(II) superoxo complex (Co1–O _ 2 _ ^ •– ^). In the presence of ^•^NO, Co1–O _ 2 _ ^ •– ^ forms a cobalt(II) peroxynitrite moiety, Co1–O _ 2 _ NO ^ – ^ (Scheme). Interestingly, the thermal decay of Co1–O _ 2 _ NO ^ – ^ at room temperature leads to the nitration of phenyl groups of the ligand at the para position to yield a unique cobalt(II)-nitrate species (Co3). Although metal-peroxynitrite-mediated nitration of phenols is known in chemistry and biology, ?−? ? ?,?,?−? ? ? nitration of aromatic hydrocarbons is elusive to date. The present study, therefore, opens up new ways for the nitration of aromatic hydrocarbons in the presence of ^•^NO/O_2_ by employing a ligand-based reservoir of electrons. This may offer significant improvements over the more conventional method of nitration in the presence of an HNO_3_/H_2_SO_4_ mixture, with regard to mild reaction conditions and green aspects by avoiding toxic catalysts and solvents. ?−? ?
Schematic Representation of the Studied Reactions of the Cobalt and Zinc Complexes
Results and Discussion
Synthesis and Characterization of the Co(II) and Zn(II) complexes
The ligand L was synthesized via a Suzuki-Miyaura coupling reaction of 2,6-bromopyridine with two equivalents of the guanidine building block (G1, Scheme S1). ^1^H and ^13^C NMR confirm the coupling of two units of G1 in 2 and 6 positions of the pyridine moiety (Figures S1, S2). Thereafter, complex Co1 was prepared under an inert atmosphere by reacting L with an equimolar amount of cobalt(II) triflate in acetonitrile at room temperature, giving rise to a greenish-red clear solution (see SI section 2). The zinc(II) analogue, Zn1, was also synthesized by using a similar procedure employing zinc(II) triflate. Single crystals of Co1 were grown by diffusing diethyl ether vapors into a concentrated acetonitrile solution of the complex at −40 °C. X-ray diffraction (XRD) analysis shows that Co1 exhibits a four-coordinate (geometry index τ_4_′ = 0.81)? distorted tetrahedral cobalt center (FigureA). It is bound by the two imine N atoms of the G1 units along with coordination from the pyridine N atom, forming an NNN-type pincer complex. The fourth coordination site is occupied by one of the two triflate anions in the unit cell, while the other one is unbound. The observed average Co–N bond distance is 1.961(3) Å, and the Co–O_triflate_ distance is 1.981(2) Å for the bound triflate. In ^1^H NMR, Co1 shows paramagnetically shifted resonances, whereas Zn1 shows a diamagnetic signal. Notably, both triflates in Co1 exist as free anions in solution, as evident from the presence of only one peak at −71.41 ppm in ^19^F NMR measurements (FigureB) corresponding to unbound triflate anions.
(A) XRD determined molecular structure of Co1, showing one bound and one unbound triflate anions per cobalt center in the solid state (one of the two molecules in the P21/C space group). Hydrogen atoms are omitted for clarity. Ellipsoids are drawn at the 50% probability level. (B) 19F NMR spectrum of Co1 in acetone-d 6.
Cyclic Voltammetry (CV)
Cyclic voltammetry measurements were performed for both complexes Co1 and Zn1 in CH_2_Cl_2_ (Figure S3). Interestingly, the CV traces were found to be nearly identical in both cases; similar nonreversible waves in the region of E 1/2 = −0.2 to 0.8 V vs Fc^+^/Fc were obtained, presumably referring to only ligand-based oxidation processes. The X-band electron paramagnetic resonance (EPR) spectrum of Co1 in a frozen CH_2_Cl_2_ solution at 13 K is consistent with the presence of a high-spin S = 3/2 (Figure, black spectrum) cobalt(II) ion in Co1. The corresponding Zn1 complex is EPR-silent, as expected.
*X-band EPR spectrum of Co1 (black) and Co1–O
2
•– (golden) in CH2Cl2 (1 mM) at 13 K. Conditions: microwave frequency 9.355 GHz; microwave power 0.016 mW; modulation amplitude 5 G.*
Oxygen Reactivity of Co1 and Zn1
Bubbling dioxygen to a 1 mM solution of Co1 in dry CH_2_Cl_2_ at −90 °C resulted in a slow but steady color change of the pale greenish solution to dark yellow (over an hour); the appearance of new absorption bands (Figure) was observed at 395 nm (ε = 2120 M^–1^ cm^–1^), 465 nm (ε = 960 M^–1^ cm^–1^), and 590 nm (ε = 460 M^–1^ cm^–1^). This oxygenated species, Co1–O _ 2 _ ^ •– ^, which is indefinitely stable at −90 °C and below, decays immediately upon increasing the temperature to room temperature to form a blue-purple solution. Crystallization of the final decay product from a concentrated tetrahydrofuran (THF) solution afforded an EPR-silent dicobalt(II) complex, Co2 (FigureA), featuring a hydroxo-bridging unit and isolated in high yield. SQUID magnetometry revealed a comparatively weak antiferromagnetic interaction of the two high-spin cobalt(II) centers with an exchange coupling constant (J) of −17.4 cm^–1^ (Figure S8). The formation of a similar hydroxo-bridged dizinc(II) complex, Zn2 (FigureB and Figures S5–S7), was obtained in high yield as a final product when Zn1 was treated with O_2_. Both Co2 (τ_4_′ = 0.83 and 0.77)? and Zn2 (τ_4_′ = 0.81 and 0.77)? exhibit two distorted tetrahedral four-coordinate Co(II) and Zn(II) centers (Co–Co and Zn–Zn distances are 3.322(2) and 3.402(3) Å, respectively), with average Co–N and Zn–N bond distances of 1.971(2) and 1.984(3) Å, respectively. The average Co–O_hydroxide_ and Zn–O_hydroxide_ distances correspond to 1.900(2) and 1.909(2) Å, respectively.
*UV–vis spectral changes associated with the formation of Co1–O
2
•– (golden) generated by bubbling O2 through a dry CH2Cl2 solution of Co1 (1 mM, black) at – 90 °C. The inset shows the time trace of the development of the bands at 395 and 455 nm.*
XRD determined molecular structures of (A) Co2 with cocrystallized tetrahydrofuran solvent molecules and (B) Zn2. Hydrogen atoms (except for the bridging μ–OH of the metalII–OH-metalII core) are omitted for clarity. Ellipsoids are drawn at the 50% probability level.
Resonance Raman (rRaman) and EPR Characterizations of Co1–O
2
•–
Resonance Raman spectroscopic measurements on Co1–O _ 2 _ ^ •– ^ at −90 °C using 407 nm excitation revealed the presence of a signal at 1201 cm^–1^, which upon ^18^O-labeling shifts to 1138 cm^–1^, indicating the formation of a superoxo species (Figures and S9). This downshift of 63 cm^–1^ is consistent with the expected shift of 68 cm^–1^ calculated by Hooke’s law for an O–O diatomic vibration. Noticeably, the (O–O) vibrational frequency observed in this case is higher than that for previously reported cobalt superoxo species, which are known to be in the typical range of 1050–1160 cm^–1^. ?−? ? ? ? ? ? The X-band EPR signal of the Co1–O _ 2 _ ^ •– ^ intermediate (Figure, golden spectrum) appears to remain mostly unaltered compared with its cobalt(II) precursor complex, indicating that the electronic structure at the metal center remains unchanged upon dioxygen activation. As the Co1 complex behaves electrochemically similar to the zinc analog, Zn1 (see above), the ligand L is anticipated to provide the required single electron to activate dioxygen to form the superoxo intermediate, Co1–O _ 2 _ ^ •– ^, at low temperature. The spins of the radical moieties L ^•+^ and O _ 2 _ ^ •– ^ must then exhibit a strong antiferromagnetic exchange interaction. Accordingly, the EPR signal of Co1–O _ 2 _ ^ •– ^, resembling that of Co1, is dominated by the cobalt(II) spin. Similar strong antiferromagnetic interactions between different radical centers, dominating over the coupling to the spin of the metal, are precedent in the literature. ?−? ? ? ? ? ? ? Possibly, the metal ions in Co1 and Zn1 behave as spectators providing Lewis acidic templates for the substrate binding site, thereby helping in polarizing O_2_, while the delocalized π-system of the guanidine-based pincer framework enables charge redistribution, driving ligand-centered redox processes critical for small-molecule activation.? The ligand-centered oxidation is further corroborated by the formation of a similar hydroxo-bridged zinc analogue, Zn2, as a final product when Zn1 is exposed to aerobic conditions. Similar to the cobalt case, Zn1–O _ 2 _ ^ •– ^ must also be involved in the reaction, which is, however, of a transient nature and could not be isolated.
*Resonance Raman (rRaman) spectra of Co1–O
2
•– in CH2Cl2 at – 90 °C (λexc = 407 nm). Black and red traces represent the spectra of the Co1–O
2
•– species generated with 16O2 and 18O2, respectively; the golden trace corresponds to the difference spectrum (18O2 – 16O2). (The asterisks mark solvent bands.).*
X-ray Absorption Spectroscopy (XAS)
X-ray absorption spectroscopy at the Co K-edge was performed on frozen solution samples of the complexes (10 mM in acetone) to assess the metal oxidation state and coordination environment (Figure). The K-edge energy of the X-ray absorption near edge structure (XANES) of Co1 indicated a cobalt(II) oxidation level, ?−? ? which was also implied by the similar K-edge energy for Co1–O _ 2 _ ^ •– ^. In agreement with the similar EPR data for both complexes, the XANES spectra further corroborate that the ligand and not the metal has become oxidized in Co1–O _ 2 _ ^ •– ^.
*(A) Normalized Co K-edge XANES spectra for Co1 (black), Co1–O
2
•– (golden), and Co1–O
2
NO
– (green) in frozen acetone solutions. The inset shows an expansion of the pre-edge region. (B) Fourier transforms (left) of the respective EXAFS spectra (right; experimental data: gray dotted line; simulations: colors as for the XANES spectra). EXAFS simulation parameters are summarized in Table S3.*
EXAFS spectra of the Co1 and Co1–O _ 2 _ ^ •– ^complexes are shown in FigureB, and their simulation parameters are summarized in Table S3. The EXAFS analysis of Co1 suggests five ligands at the metal in an acetone solution, i.e., three Co–N_average_ (2.01 Å) from L and two Co–N/O (2.14 Å) bonds, possibly from triflate/solvent ligands, while the XRD structure shows only one triflate^–^ ligand at cobalt and an overall ca. 0.08 Å shorter Co–N/O bond length. The EXAFS spectrum of Co1–O _ 2 _ ^ •– ^ was similarly well simulated with a five-coordinate Co center with three Co–N (1.98 Å) from L and two Co–N/O (2.13 Å) bonds from O_2_ ^•–^. The slight changes in bond lengths may be rationalized by the exchange of a solvent ligand by another species, i.e., the superoxide ligand.
DFT Calculations
DFT calculations were performed to get insight into the nature of the superoxo intermediate Co1–O _ 2 _ ^ •– ^. Geometry optimizations of a variety of starting structures (see SI section 8) yielded stable superoxo complexes only for the side-on binding mode. In contrast, the end-on binding mode always led to structures with significantly shorter O–O distances corresponding to a dioxygen unit often dissociated from the Co center. Broken-symmetry (BS) calculations for total spin states S_t_ = 3/2 (S_1_ = 2, S_2_ = 1/2) and S_t_ = 1/2 (S_1_ = 3/2, S_2_ = 1) did not converge to true BS solutions, but yielded the same structures and wave functions as the corresponding S = 3/2 and 1/2 calculations, respectively, without using the BS approach. Consistent with the experimental data, the minimum-energy structure was found for the spin state of 3/2 (Figure, Table S1), which is 2.0 and 3.1 kcal/mol lower in energy than those of the S = 5/2 and 1/2 structures, respectively. However, its spin density (Figure S10) as well as inspection of the frontier orbitals indicate an **LCo(III)-O_2_ ^•–^ ** electronic configuration, with an intermediate spin (S = 1) Co(III) center ferromagnetically coupled to the superoxo unit and no oxidation of the ligand L. This is in contrast to the results from CV, XANES, and EPR (which suggest antiferromagnetic interaction of the superoxo and ligand radicals). The average Co–N bond distance is 1.896 Å, the Co–O distances are 1.880 and 1.963 Å, and the O–O distance is 1.296 Å. The calculated vibrational frequency of the O–O stretching mode is 1198 cm^–1^, downshifting by 69 cm^–1^ upon ^18^O-labeling. This is in excellent agreement with the experimental frequencies and strongly supports the side-on superoxo model. In summary, the DFT approach apparently does not provide the correct description of the electronic structure of the Co1–O _ 2 _ ^ •– ^ intermediate. Nevertheless, due to the exclusive stabilization of the side-on binding mode for the superoxo ligand and the agreement of experimental and calculated O–O stretching energies, we tentatively assign Co1–O _ 2 _ ^ •– ^ as a side-on superoxo complex.
*Minimum-energy (St = 3/2) DFT structure for Co1–O
2
•– . (Hydrogen atoms are omitted for the sake of clarity.).*
Reactivity with Nitric Oxide
Co1–O _ 2 _ ^ •– ^ instantaneously reacts with ^•^NO at −90 °C to generate a forest green colored species, Co1–O _ 2 _ NO ^ – ^, with the concomitant formation of an intense broad band at 635 nm (ε = 660 M^–1^cm^–1^) in the UV–vis absorption spectrum (Figure), similar to characteristic ligand-to-metal charge transfer bands of typical peroxo species. ?−? ? ? ? ?
*Formation of Co1–O
2
NO
– (green trace) by reaction of excess •NO gas with Co1–O
2
•– (1 mM in CH2Cl2, golden trace; cell path-length: 0.5 cm) as monitored by UV–vis spectroscopy at – 90 °C.*
In the rRaman spectra of Co1–O _ 2 _ NO ^ – ^ (Figure), the O–O stretch of Co1–O _ 2 _ ^ •– ^ at 1201 cm^–1^ completely disappeared and two new ^16/18^O isotope sensitive vibrational modes were observed at 808 and 1598 cm^–1^, which upon ^18^O-labeling shift to 773 and 1595 cm^–1^, respectively. These bands are tentatively assigned to CoO-ONO and CoO_2_NO vibrational modes, particularly in view of the similar isotopic shift pattern as in the literature. ?,? X-band EPR measurement (13 K) of the solution of Co1–O _ 2 _ NO ^ – ^ frozen in liquid N_2_ right after the addition of ^•^NO to Co1–O _ 2 _ ^ •– ^ at −90 °C shows that the S = 3/2 signal from high-spin cobalt(II) in Co1–O _ 2 _ ^ •– ^ vanishes entirely, resulting in the formation of an EPR-silent species (FigureA). This observation can be ascribed to spin coupling between the high-spin cobalt(II) core and the ligand radical L ^•+^, resulting in an integer spin (S = 1 or 2) for Co1–O _ 2 _ NO ^ – ^. A slightly higher K-edge energy (FigureA, green) was observed for Co1–O _ 2 _ NO ^ – ^, which may suggest the formation of cobalt(III) in part of the sample. However, the largely increased pre-edge amplitude and the broader rising edge of Co1–O _ 2 _ NO ^ – ^ rather were indicative of a diminished coordination symmetry at the metal site, so that we assign Co(II) also to Co1–O _ 2 _ NO ^ – ^. The EXAFS spectrum of Co1–O _ 2 _ NO ^ – ^ (FigureB, green) revealed only four ligands at the metal (i.e., three Co–N and one Co–N/O bonds), furthermore resulting in overall almost 0.1 Å shorter bond lengths, with the shortest bond at ca. 1.8–1.9 Å. In addition, the simulation suggested a further light atom in the second coordination sphere at a distance of ca. 2.45–2.70 Å to the metal, which was seemingly absent in the precursor complexes Co1 and Co1–O _ 2 _ ^ •– ^. This atom may stem from a diatomic (or higher atomic) N/O-containing ligand bound to the cobalt center, such as peroxynitrite (^ – ^O_2_NO). Binding of this ligand seems to cause the loss of the fifth ligand from the metal.
*rRaman spectra of Co1–O
2
NO
– in CH2Cl2 at – 90 °C (λexc = 407 nm). Black and red spectra refer to Co1–O
2
NO
– generated from the reaction of •NO with Co1-
16
O
2
•– and Co1-
18
O
2
•– , respectively. (The asterisks mark solvent bands.).*
Although Co1–O _ 2 _ NO ^ – ^ is stable at −90 °C for more than an hour, it undergoes a thermal decay at higher temperatures, presumably by O–O bond homolysis to form L^•+^Co(II)-oxyl and ^•^NO_2_ radical species, indicated by X-band EPR measurements on a reaction aliquot sample taken immediately after the reaction mixture has reached 0 °C from −90 °C during the process of warming up. The EPR-silent solution of Co1–O _ 2 _ NO ^ – ^ transforms (FigureA,B) to a solution that exhibits an S = 3/2 signal corresponding to L^•+^Co(II)-oxyl (10% yield based on Co) and a radical-based signal at g = 2 with dominant ^14^N-hyperfine splitting. The signal shape and turning points are characteristics of the ^•^NO_2_ radical as previously reported, ?−? ? ? which are confirmed by the * g
- = [2.0043, 1.9901, 2.0008] and hyperfine (* A *) [142, 134, 188] MHz tensor components obtained from spectral simulations (Figure S11). The formation of ^•^NO_2_ is further confirmed by the appearance of a reddish-brown gas in the reaction vessel upon reaching room temperature (Figures S12–S13). The signal corresponding to the ^•^NO_2_ radical eventually disappears after reaching room temperature (FigureB,C), and the S = 3/2 cobalt(II) signal persists with approximately 80% depleted intensity compared to the starting Co1 complex. Extracting the final decay product with tetrahydrofuran revealed the formation of two different cobalt complexes, Co2 and Co3, in 62 and 21% yields, respectively, with respect to Co1. The different solubilities of Co3 and Co2 in THF enabled their separation, with Co3 subsequently being isolated by recrystallization of the remaining crude product from acetonitrile. The crystal structure of Co3 (Figure) exhibits a six-coordinate cobalt(II) center with two anions, one triflate and one nitrate, bound to it. The average Co–N distance is 2.084(14) Å, and Co–O_triflate_ is 2.189(10) Å. Interestingly, the generation of Co3 is accompanied by concomitant nitration of the two phenyl rings in the ligand backbone, selectively at the para positions. When the thermal decay of Co1–O _ 2 _ NO ^ – ^ was performed in the presence of an external substrate like anisole, both ortho and para-nitro anisole were obtained in 37.2% yield each, with respect to the peroxynitrate species (see SI section 2 and Figure S14).
XRD determined molecular structure of Co3. (Hydrogen atoms are omitted for clarity. Ellipsoids are drawn at the 25% probability level.).
*Monitoring of the thermal decomposition of Co1–O
2
NO in CH2Cl2 (1 mM) by X-band EPR spectroscopy: (A) initial spectra corresponding to Co1–O
2
NO at – 90 °C; (B) transient formation of L•+Co(II)-oxyl (10% yield) and •NO2 as observed from a reaction mixture aliquot taken immediately after warming to 0 °C; and (C) the final formation of Co3 (20% yield) at room temperature. Conditions: microwave frequency 9.355 GHz, microwave power 0.016 mW, modulation amplitude 5 G, and temperature 13 K.*
Thus, L^•+^Co(II)-oxyl and ^•^NO_2_, formed upon O–O bond homolysis of Co1–O _ 2 _ NO ^ – ^, may either rearrange to form a nitrate anion, or the ^•^NO_2_ radical in the presence of L^•+^Co(II)-oxyl may be responsible for the nitration of the phenyl rings in the para positions of the ligand. The plausible reaction sequence for the generation of Co3 and Co2 from the thermal decay of Co1–O _ 2 _ NO ^ – ^ is shown in Scheme. Consistent with the proposed mechanism, the decay of Co1–O _ 2 _ NO ^ – ^ is found to be dependent on cobalt concentration (Figure S12), thereby suggesting an intermolecular mechanism. In addition, dioxygen is detected by gas chromatography (Figure S13). Notably, thermal and photochemical nitration of aromatic hydrocarbons with ^•^NO_2_ has been reported as an alternative to the usual electrophilic substitution pathway for aromatic nitration reactions involving nitrosonium cations (NO_2_ ^+^). ?,? The present study establishes a plausible synergistic role of metal-oxo and ^•^NO_2_, obtained from the decay of metal-peroxynitrites, in mediating aromatic nitration reactions under mild conditions in a greener way by avoiding toxic catalysts and solvents. These aspects will be investigated in detail in future studies.
*Proposed Reaction Scheme for the Thermal Decomposition of Co1–O
2
NO to the (L*)CoII Moiety in Co3 and the L-CoII–OH-CoIIL Unit in Co2*
Conclusions
Herein, we demonstrated activation of O_2_ via a ligand-based redox process to generate an unusual Co(II) superoxo species. The superoxo complex was characterized by EPR, XAS, and rRaman spectroscopic techniques to validate the proposed ligand-based reactivity pathway. This complex can react with ^•^NO to form a Co peroxynitrite species, which performs an unprecedented intermolecular aromatic nitration reaction. The observed ligand-based electron transfer to NO/O_2_ may be related to that in biological systems proposed to utilize a similar cobalt-peroxynitrite intermediate.? This work represents an important step forward in leveraging ligand-based redox for inexpensive and benign aromatic nitration reactions with cobalt.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Huie R. E.Padmaja S.The reaction of NO with superoxide Free Radical Res. Commun.199318419519910.3109/107157693091458688396550 · doi ↗ · pubmed ↗
- 2Mittal, M. ; Siddiqui, Mr. Fau. ; Tran, K. ; Tran, K. Fau. ; Reddy, S. P. ; Reddy, Sp. Fau. ; Malik, A. B. ; Malik, A. B. , Reactive oxygen species in inflammation and tissue injury. (1557–7716 (Electronic)).10.1089/ars.2012.5149 PMC 392901023991888 · doi ↗ · pubmed ↗
- 3Buonocore G.Perrone S.Tataranno M. L.Oxygen toxicity: chemistry and biology of reactive oxygen species Semin. Fetal. Neonatal. Med.201015418619010.1016/j.siny.2010.04.00320494636 · doi ↗ · pubmed ↗
- 4Rauf A.Khalil A. A.Awadallah S.Khan S. A.Abu-Izneid T.Kamran M.Hemeg H. A.Mubarak M. S.Khalid A.Wilairatana P.Reactive oxygen species in biological systems: Pathways, associated diseases, and potential inhibitorsA review Food Sci. Nutr.202412267569310.1002/fsn 3.378438370049 PMC 10867483 · doi ↗ · pubmed ↗
- 5Farah C.Michel L. Y. M.Balligand J.-L.Nitric oxide signalling in cardiovascular health and disease Nat. Rev. Cardiol.201815529231610.1038/nrcardio.2017.22429388567 · doi ↗ · pubmed ↗
- 6Gardner, P. R. ; Gardner, A. F. ; Martin, L. A. ; Martin, L. F. ; Salzman, A. L. ; Salzman, A. L. , Nitric oxide dioxygenase: an enzymic function for flavohemoglobin. (0027–8424 (Print)).10.1073/pnas.95.18.10378 PMC 279029724711 · doi ↗ · pubmed ↗
- 7Tocheva E. I.Rosell F. I.Mauk A. G.Murphy M. E. P.Side-On Copper-Nitrosyl Coordination by Nitrite Reductase Science 2004304567286787010.1126/science.109510915131305 · doi ↗ · pubmed ↗
- 8Gardner P. R.Nitric oxide dioxygenase function and mechanism of flavohemoglobin, hemoglobin, myoglobin and their associated reductases J. Inorg. Biochem.200599124726610.1016/j.jinorgbio.2004.10.00315598505 · doi ↗ · pubmed ↗