A Case of Polymerase Proofreading‐Associated Polyposis: Challenges in Genetic Diagnosis
Haruka Ito, Akiko Chino, Arisa Ueki, Keika Kaneko, Shoichi Saito

TL;DR
This paper presents a rare case of PPAP in Japan, emphasizing the challenges in diagnosing this hereditary condition and the importance of genetic testing.
Contribution
The paper contributes a rare Japanese case of PPAP and highlights the need for multi-gene panel testing and variant reinterpretation for accurate diagnosis.
Findings
A 50-year-old woman was diagnosed with PPAP after comprehensive genetic testing revealed a likely pathogenic POLE variant.
The case illustrates the diagnostic challenges of PPAP due to its similarity to other polyposis syndromes.
The patient's complex cancer history underscores the importance of multidisciplinary care and continued surveillance in PPAP.
Abstract
Polymerase proofreading‐associated polyposis (PPAP) is a rare autosomal dominant hereditary syndrome caused by germline pathogenic variants in the POLE or POLD1 genes. It is clinically similar to familial adenomatous polyposis (FAP) and Lynch syndrome, making diagnosis difficult. Although the number of reported cases is increasing globally, PPAP remains underrecognized, particularly in Japan. Accurate diagnosis often requires comprehensive genetic testing, including multi‐gene panel analysis and variant reinterpretation. We report a rare case of PPAP in a 50‐year‐old woman with a complex clinical history involving multiple primary malignancies. The patient developed ovarian cancer in her 20s, followed by endometrial and contralateral ovarian cancers in her 30s. She was also diagnosed with early‐stage colorectal cancer and polyposis, for which she underwent total colectomy with…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
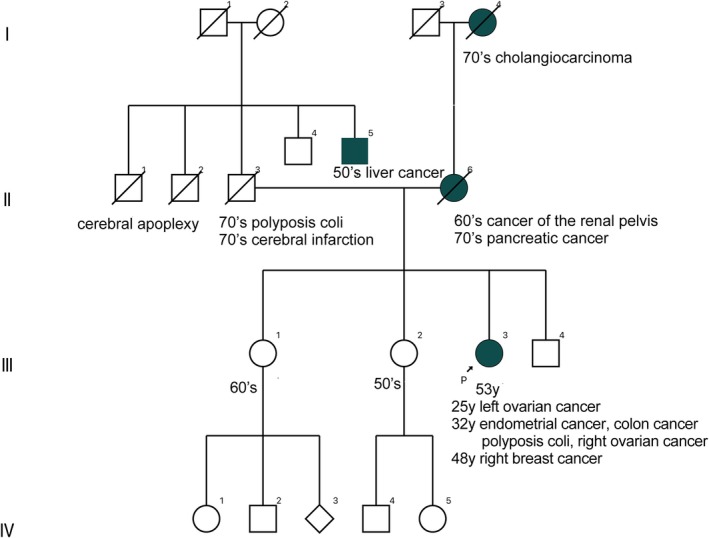 FIGURE 1
FIGURE 1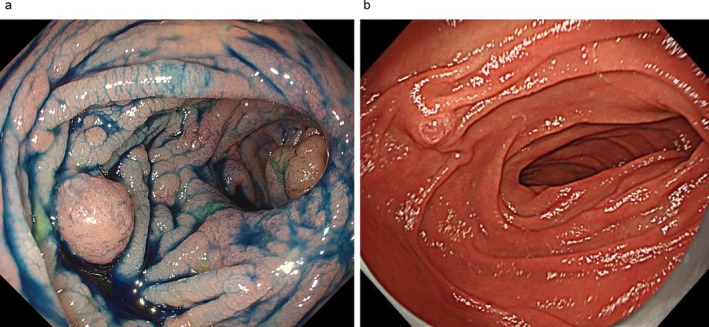 FIGURE 2
FIGURE 2| Year/reporter | Age | Sex | Age at initial cancer diagnosis | Age at genetic diagnosis | Initial cancer type | Metachronous cancers | Causative gene | Cancer types among family |
|---|---|---|---|---|---|---|---|---|
| Yamaguchi et al. [ | 68 | M | 68 | Unknown |
Colon cancer Polyposis coli | Unknown |
|
Thyroid cancer Gastric cancer Pancreatic cancer |
| Ito et al. [ | 49 | F | 49 | Unknown | Polyposis coli |
Colon cancer Endometrial cancer |
| Brain tumor |
| Miyazaki et al. [ | 43 | F | 29 | Unknown | Colon cancer |
Duodenal tumor Endometrial cancer Thyroid cancer |
|
Brain tumor Thyroid cancer |
| 2024 Our case | 53 | F | 25 | 53 | Ovarian cancer |
Colon cancer Endometrial cancer Breast cancer |
| None |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic factors in colorectal cancer · Cancer Genomics and Diagnostics · Colorectal Cancer Treatments and Studies
Introduction
1
Polymerase proofreading‐associated polyposis (PPAP) is a rare autosomal dominant hereditary syndrome caused by germline pathogenic variants in the POLE or POLD1 genes. These genes encode DNA polymerases responsible for proofreading and correcting errors during DNA replication, and their dysfunction leads to an increased risk of colorectal adenomas and various malignancies. PPAP shares overlapping clinical features with familial adenomatous polyposis (FAP) and Lynch syndrome, making accurate diagnosis challenging. Although cases have been increasingly reported worldwide, PPAP remains underrecognized, particularly in Japan. We present a detailed case of PPAP in a 50‐year‐old woman with a complex cancer history, highlighting the diagnostic challenges and the role of multi‐gene panel testing in establishing the diagnosis.
Case Report
2
We report a case of PPAP in a 50‐year‐old woman. PPAP is an autosomal dominant hereditary disorder caused by germline pathogenic variants in the POLE or POLD1 genes, which are responsible for correcting errors during DNA replication. Patients typically develop several dozen colorectal adenomas. Extra‐colonic manifestations have been reported to vary depending on the affected gene: POLE‐related PPAP is associated with duodenal adenomas or carcinomas and brain abscesses, while POLD1‐related PPAP has been linked to endometrial cancer, breast cancer, and brain tumors [1]. Because PPAP can present with clinical features similar to those of FAP and Lynch syndrome, genetic testing is essential for accurate diagnosis and differentiation.
She had been diagnosed with left ovarian cancer in her 20s and underwent a left adnexectomy. In her 30s, she was diagnosed with endometrial cancer and right ovarian cancer, for which she underwent total hysterectomy, right adnexectomy, omentectomy, and pelvic and para‐aortic lymphadenectomy. Around the same time, early‐stage colon cancer and adenomatous polyposis were also identified, and she underwent total colectomy with ileorectal anastomosis (IRA). Based on the clinical course, FAP of the colon was suspected, and genetic counseling was recommended first. The patient's family history revealed that her mother had pancreatic cancer, and her maternal grandfather had bile duct cancer; however, there was no evidence of colorectal cancer or gastrointestinal polyposis in the family history (Figure 1). In 200X, genetic counseling was conducted at our hospital, and germline pathogenic variants (GPVs) in APC and MUTYH were analyzed through genetic tests. However, no GPVs were found in APC, and a variant of uncertain significance (VUS) was found in MUTYH. In 200X + 2, immunohistochemical staining (IHC) of colorectal cancer showed loss of MSH2 and MSH6 proteins; therefore, genetic testing targeting MSH2 and MSH6 was performed, but GPVs were not reported. Although the patient's family history raised suspicion for Lynch syndrome, no pathogenic variants were identified in the associated genes, and the diagnosis was therefore considered unlikely. Afterward, the patient developed right breast cancer in her 40s. In 200X + 16, multi‐gene panel testing (MGPT) targeting 22 genes, including BRCA1/2 and POLE, was performed, which revealed a VUS in POLE but did not lead to a definite diagnosis. During continued surveillance as a high‐risk case, the interpretation of the variants was revised as additional scientific evidence emerged following the initial analysis, and our institution, which had ordered the test, was subsequently notified.
There are no cases of colorectal cancer or gastrointestinal polyposis in patient's family. This case is shown as III‐3.
In 2020, this variant was reported in a female individual with multiple early‐onset malignancies, including colorectal, ovarian, and endometrial cancer [2]. Moreover, they reported functional evidence from a yeast model indicating a possible impact on protein function. It is presumed that, based on this publication, the testing laboratory reclassified the variant as likely pathogenic. As a result, 5 years after the MGPT, an amended report was issued: VUS in POLE was reclassified as likely pathogenic, and the VUS in MUTYH was reinterpreted as likely benign. Based on this re‐evaluation, in combination with the patient's clinical phenotype, PPAP's diagnosis was established. The colorectal feature in this patient was the absence of polyps in the early years after IRA, the appearance of solitary polyps that were removed annually in her 30s, and a gradual increase in the number of polyps from her 40s (Figure 2a). In the residual rectum, she developed intramucosal carcinoma twice at 200X + 11 and 200X + 21 years. Furthermore, a tubulovillous adenoma > 30 mm in diameter developed within the J‐pouch at the anastomosis. Regarding upper gastrointestinal diseases, no polyposis of the gastric fundus or duodenal polyps was observed (Figure 2b).
(a) Adenomatous polyposis of the remaining rectum seen on colonoscopy. (b) Descending part of the duodenum. In all cases, a genetic diagnosis was reached through the WGS or MGPT. The patient selected the Colorectal Cancer Panel (VistaSeq), which targets 22 genes associated with colorectal cancers.
Discussion
3
The case reports of PPAP searched in the Central Journal of Medicine and PubMed from 2013 to 2024 revealed four reported cases, including our case, in Japan [3, 4, 5]. (Table 1). Of the three patients other than our patient, one was diagnosed with GPV in POLE, and two were diagnosed with GPVs in POLD1; all of them had a history of colon cancer. All other patients had a family history of PPAP.
Recommendations for the surveillance of PPAP have not yet been clearly established, as the number of reported cases remains limited, and available evidence is scarce. Consequently, current surveillance strategies are often guided by those used for FAP and Lynch syndrome. Based on the author's clinical perspective, an annual colonoscopy is advisable, with adjustments to the surveillance interval made according to the number and histological characteristics of polyps. For upper gastrointestinal tract evaluation, periodic upper endoscopy every 1–3 years may be appropriate, especially in patients with POLE mutations, given reports of duodenal adenomas and carcinomas in this population. For female patients, annual gynecological examinations and transvaginal ultrasonography are recommended, along with endometrial cytology, as needed. Although these strategies are not yet standardized, they represent the author's current approach to the clinical management of PPAP.
MGPT has proven to be a valuable tool for diagnosing adenomatous polyposis, a condition that presents a significant challenge in genetic assessment. This patient underwent prolonged consultations with medical professionals, including genetic counselors, to ascertain a specific phenotype, facilitating subsequent communication and collaboration.
Ethics Statement
The authors have nothing to report.
Consent
Informed consent was obtained from this participant.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1F. Bellido , M. Pineda , G. Aiza , et al., “POLE and POLD 1 Mutations in 529 Kindred With Familial Colorectal Cancer and/or Polyposis: Review of Reported Cases and Recommendations for Genetic Testing and Surveillance,” Genetics in Medicine 18 (2016): 325–332.26133394 10.1038/gim.2015.75PMC 4823640 · doi ↗ · pubmed ↗
- 2N. Hamzaoui , F. Alarcon , N. Leulliot , et al., “Genetic, Structural, and Functional Characterization of POLE Polymerase Proofreading Variants Allows Cancer Risk Prediction,” Genetics in Medicine 22 (2020): 1533–1541.32424176 10.1038/s 41436-020-0828-z · doi ↗ · pubmed ↗
- 3K. Yamaguchi , E. Shimizu , and R. Yamaguchi , “Development of an MSI‐Positive Colon Tumor With Aberrant DNA Methylation in a PPAP Patient,” Journal of Human Genetics 64 (2019): 729–740.31089268 10.1038/s 10038-019-0611-7 · doi ↗ · pubmed ↗
- 4T. Ito , T. Nomizu , H. Eguchi , et al., “The First Case Report of Polymerase Proofreading‐Associated Polyposis in POLD 1 Variant, c.1433 G>A p.S 478N, in Japan,” Japanese Journal of Clinical Oncology 50 (2020): 1080–1083.32548621 10.1093/jjco/hyaa 090 · doi ↗ · pubmed ↗
- 5H. Miyazaki , O. Dohi , E. Maeda , et al., “Multiple Duodenal Epithelial Tumors in a Patient With Polymerase Proofreading‐Associated Polyposis in POLE Variant,” Clinical Journal of Gastroenterology 17 (2024): 425–428.38386255 10.1007/s 12328-024-01922-1 · doi ↗ · pubmed ↗