Missense variants in SLC9A6 cause partial epilepsy without neurodevelopmental delay
Jun-Ping Jiao, Hong-Wei Zhang, Xi-Zhong Zhou, Shu-Juan Tian, Li Gao, Bing-Mei Li, Jun-Xia Luo, Jie Wang, Song Lan, Bin Li, Wei-Ping Liao

TL;DR
Missense variants in the SLC9A6 gene are linked to mild epilepsy without severe developmental issues.
Contribution
Identifies a genotype-phenotype correlation and sub-regional effects of SLC9A6 variants in epilepsy.
Findings
Missense variants in SLC9A6 are associated with mild partial epilepsy.
Variants in specific regions of SLC9A6 correlate with milder symptoms compared to null variants.
Previously reported missense variants outside key regions are linked to less severe outcomes.
Abstract
The SLC9A6 gene encodes a monovalent sodium-selective sodium/hydrogen exchanger that is essential in regulating endosomal PH and volume. SLC9A6 variants are associated with Christianson Syndrome, a severe neurodevelopmental disorder that is accompanied by seizures. It is unknown whether SLC9A6 variants are associated with milder phenotypes. Trio-based whole-exome sequencing was performed in unrelated cases (families) with epilepsy without acquired causes. Previously reported SLC9A6 variants were reviewed to analyze the mechanism underlying phenotype variations. Five hemizygous variants, including three null and two missense variants, were identified in five males. All the variants were absent in the gnomAD-all populations and the missense variants were predicted to be damaging by multiple in silico tools. The three patients with null variants presented with refractory epilepsies and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
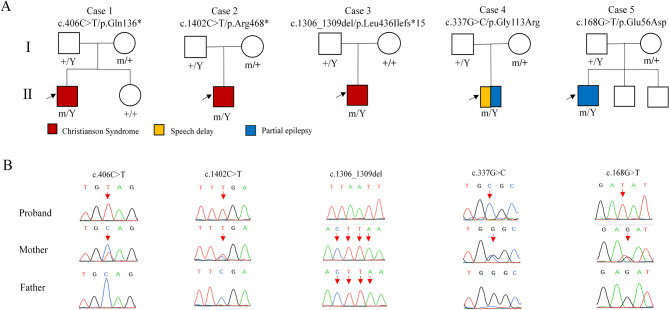 Figure 1
Figure 1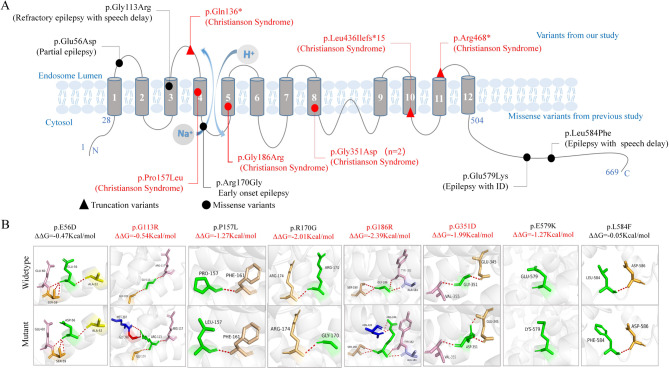 Figure 2
Figure 2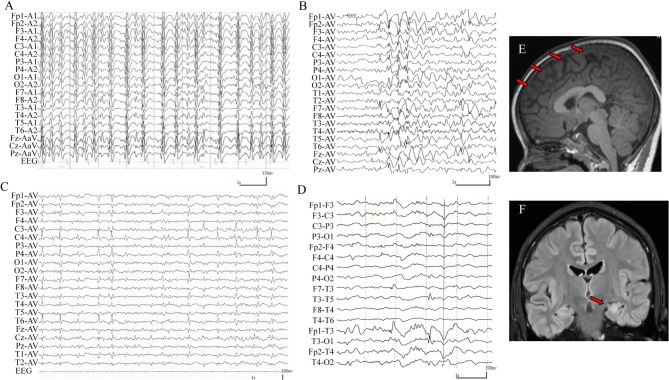 Figure 3
Figure 3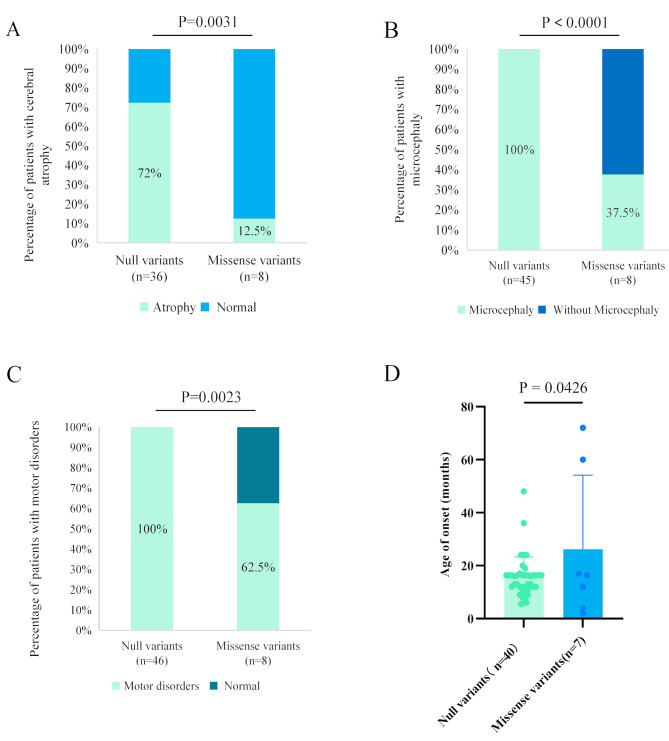 Figure 4
Figure 4- —http://dx.doi.org/10.13039/501100001809National Natural Science Foundation of China
- —Guangdong Basic and Applied Basic Research Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAmino Acid Enzymes and Metabolism · Cystic Fibrosis Research Advances · Genomics and Rare Diseases
Introduction
The SLC9A6 gene (OMIM* 300231), also known as NHE6 gene, is located on chromosome Xq26.3 and encodes a monovalent sodium-selective sodium/hydrogen exchanger (NHE) [1, 2]. The SLC9A6 protein is mainly expressed in human brain tissues [1, 3]. It is found in the membranes of intracellular organelles such as mitochondria and endosomes, playing an essential role in regulating endosomal pH and volume [4–7]. Homozygous KO mice show postnatal lethality [8, 9] suggesting an essential role of SLC9A6 in neuronal development.
Previously, SLC9A6 variants have been reported to be associated with X-linked syndromic intellectual developmental disorder (MRXSCH, OMIM # 300243), as known as Christianson Syndrome (CS) [10–14]. CS is characterized by early-onset refractory epilepsy, severe intellectual disability (ID), and global developmental delay (GDD) [12, 13, 15, 16]. It is unknown whether SLC9A6 variants are associated with pure epilepsies, and the mechanisms underlying phenotypical variations remain elusive [17].
In the present study, trio-based whole exome sequencing (WES) was performed in cases (families) of epilepsy without acquired causes. SLC9A6 hemizygous variants, including three null and two missense variants, were identified in five males with epilepsy. We further reviewed all previously reported SLC9A6 variants and analyzed their molecular heterogeneity, aiming to explore the mechanism underlying phenotypical variation.
Materials and methods
Participants
The patients in this study were recruited from a multicenter cohort via the China Epilepsy Project 1.0 (https://epg1.cn/), which included fifteen cohorts subclassified by clinical phenotypes. Detailed clinical information, including clinical phenotypes, age at onset of seizures, seizure types, duration and frequency of seizures, family history, treatment status, prognosis, routine and neurological physical examinations, as well as brain magnetic resonance imaging (MRI), was collected. Electroencephalogram (EEG) monitoring, including hyperventilation, intermittent photic stimulation, eye-opening test, and sleep recording, was analyzed by at least two qualified EEG technicians. The diagnosis of epilepsy and seizure classification followed the criteria established by the International League Against Epilepsy (ILAE) [18–22]. Patients with acquired causes of epilepsy were excluded.
WES
The study collected blood samples from both the probands and their parents, and genomic DNA was extracted from the peripheral blood using the Qiagen Flexi Gene DNA kit (manufactured by Qiagen, Hilden, Germany) in accordance with the manufacturer’s instructions. A trio-based WES was performed on the MGI 2000 platform by BGI-Shenzhen. The WES consisted of paired-end reads with 100 bp sequencing, resulting in an average depth of coverage of 100-150X and a target region coverage exceeding 98%. The deep sequencing data were aligned to the GRCh37 build (hg19) reference genome. Following this alignment, single-nucleotide variants and insertion/deletion variants were identified and annotated using the Genome Analysis Tool Kit as previously reported [23, 24].
Genetic analysis
To identify potential pathogenic variants in each individual case, a tailored strategy was employed. Initially, rare variants with a minor allele frequency below 0.005 in publicly available databases such as the 1,000 Genomes Projects, Exome Aggregation Consortium, and gnomAD were firstly prioritized. Then, potentially disease-causing variants, such as canonical splicing, nonsense, frameshift, in-frame indels, missense, and initiation codon variants, were selected for further analysis. Subsequently, stratified minor allele frequency (MAF) criteria were applied: de novo, hemizygous, and homozygous variants should not be found in control populations in gnomAD. For biallelic variants, the product of the frequencies of the two alleles in gnomAD should be less than 1 × 10^− 6^, which is significantly lower than the expected probability of such an occurrence in the current gnomAD population (1/141456 = 7 × 10^− 6^). Ultimately, the candidate variants were refined through the application of stringent criteria sourced from the Genetic Dependence & Pathogenicity Database (www.gdap.org.cn), encompassing four pivotal facts of the gene profiles:
-
Tissue-specific expression: a prerequisite for a gene to be considered epilepsy-causing is its expression within the brain (inclusion criterion). Additionally, alternative explanations, such as ectopic expression or remote toxic effects stemming from abnormal metabolic byproducts, were also taken into consideration.
-
Exclusion of conflicting gene-disease associations: genes that have been definitively linked to diseases that are incompatible with the epilepsy phenotype were systematically excluded from further consideration (exclusion criterion).
-
Gene intolerance to variants: the probability of a gene being intolerant to heterozygous or homozygous loss-of-function variants (pLI/pRec) was evaluated. Specifically, genes with a pLI score of 0.9 or higher, harboring dominant variants, and those with both pLI and pRec scores of 0.9 or higher, along with a pNull score of 0.1 or lower in the context of recessive variants, were given particular attention.
-
Phenotypic consequences of genetic manipulation: whether genetic knockout or knockdown experiments resulted in brain-related phenotypes was explored as a way of validating the pathogenicity of genes.
After applying the filtering criteria, genes with recurrent variants were chosen for further investigation to elucidate their specific gene-disease associations. One of the candidate genes that emerged from our analysis was SLC9A6. Sanger sequencing was employed to validate our findings and confirm the origin of the identified variants. All the identified variants within the SLC9A6 gene in this study were annotated using the reference transcript NM_006359.3.
Damaging effects of SLC9A6 variants
We used nine commonly used in silico tools, including SIFT, FATHMM_MKL, Mutation-Taster, CADD, Geno-Canyon, fitCons, M_CAP, GERP++, and SiPhy, to predict the impacts of missense variants. The results were obtained from the VarCards database (http://genemed.tech/varcards), an integrated genetic and clinical database for coding variants in the human genome.
To predict the impact of missense variants on molecular structure, we utilized the SWISS-MODEL to conduct protein modeling. The three-dimensional protein structure and hydrogen bonding alterations of missense variants were visualized and analyzed by PyMOL 2.6. The changes of the protein stability were assessed using the free energy stability change (DDG, Kcal/mol) value. Amino acids were annotated to the reference protein NP_006350.
Exploring of mechanism underlying phenotype variations
To reveal the underlying mechanism of phenotype variations, we first reviewed all the relevant literature about SLC9A6 variants reported previously. All variants were obtained from PubMed [4] and the Human Gene Mutation Database [5] up until March 2023 (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php). Variants, such as nonsense, frameshift, splicing variants in classical soil 1 or 2, start codon, single or multiple exon deletion variants were treated as null variants. Then, we explored (1) genotype-phenotype correlation by comparison of proportions of patients with cerebral atrophy, microcephaly, and motor disorders in patients with null variants and missense variants; and (2) sub-molecular effect, the locations and damaging effects of previously reported missense variants and their phenotype severity.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software 10.0. The data are expressed as mean ± SEM. Statistical comparisons were conducted using Student’s t-tests and Chi-square tests, with a significance level set at p < 0.05.
Results
Identification of SLC9A6 variants
Five hemizygous SLC9A6 variants, including three null (c.406 C > T/p.Q136*, c.1402 C > T/p.R468*, and c.1306_1309del/ p.L436Ifs15) and two missense variants (c.168G > T/p.E56D and c.337G > C/p.G113R), were identified in five unrelated individuals (Fig. 1; Table 1). The variant c.1306_1309del/p.L436Ifs15 arose de novo and the other variants were inherited from unaffected mothers, consistent with an X-linked recessive inheritance pattern.
Fig. 1. Genetic characterization of the cases with SLC9A6 variants. (A) Pedigrees of the cases with SLC9A6 variants and their corresponding phenotypes. (B) DNA sequence chromatogram of the SLC9A6 variants. Arrows indicated the positions of the variants
Table 1. Clinical features of the cases with SLC9A6 variantsCase IDVariant(NM_006359.3)GenderAgeSeizure onsetSeizure typeASMsSeizure outcomeEEGCerebral atrophyIDMicrocephalyMotor developmentStrabismusDiagnosis1c.406 C > T/p.Gln136Male8 y9 mTS, GTCS, AS,(6–7 times/ d)VPA,LEV,TPMLTG,CLB2–3 times/ d~1/ mESES (5 y)YesSevereYesSpeech delay, unable to standYesCS2c.1402 C > T/p.Arg468Male3 y1 yGTCS (1–2 times/d),SPS (1–2 times/ d)LEV,PER2–4 times/ mDiffuse discharges (1 y)Yes (1 y)SevereYesUnable to stand and talkYesCS3c.1306_1309del/p.Leu436Ilefs*15Male5 y1 y 7 mCPS, GTCSMultiple ASMsNANAYes (23 m)SevereYesUnable to stand and talkYesCS4c.337G > C/p.Gly113ArgMale8 y5 yCPS(5–6 times/ m)PER, LEVCPS4–5 times/ yBilateral central, parietal, and temporal discharges independently (8y)No (5 y)NoNoSpeech delayNoPartial epilepsy5c.168G > T/p.Glu56AspMale17y6 yCPS (5-9 times/ d)LTG, VPASeizure free for 18 mLeft temporal discharges at 11 y and is normal at 13 y/15 y/ 17 yNo atrophy, hippocampal sclerosis (15 y)NoNoNormalNoPartial epilepsyAbbreviations: AS, absence seizure; CLB, clobazam; CPS, complex partial seizure; CS, Christianson syndrome; d, day; ESES, electrical status epilepticus in slow-wave sleep; GDD, global developmental delay; GTCS, generalized tonic-clonic seizure; LEV, levetiracetam; LTG, lamotrigine; m, month; NA, not available; OFC, occipital frontal circumference; PER, perampanel; SPS, simple partial seizure; TPM, topiramate; TS, tonic seizure; VPA, valproic acid; y, year
All the three hemizygous null variants (PVS1) were not present in gnomAD-all populations (PM2) (Table 2). The c.406 C > T/p.Q136* and c.1402 C > T/p.R468* were predicted to be damaging by in silico tools (PP3). The c.1402 C > T/p.R468* was a previously reported pathogenic variant (PS1). The c.1306_1309del/ p.L436Ifs*15 was de novo origin (PS2). According to ACMG guidelines, the three null variants were evaluated to be “Pathogenic”.
Table 2. Allele frequencies and bioinformatics analysis of the SLC9A6 variants identified in this studyCaseNocDNA change(NM_022168.4)Protein changeInheritanceMAFIn silico missense predictionACMG (scoring)gnomADgnomAD- EASSIFTFATHMM_MKLMutation-TasterCADDGenoCanyonfitConsM_CAPGERP++SiPhy1c.406 C > Tp.Gln136Maternal---D(0.913)D(1)D(37)D(1)--C(5.07)C(16.698) P (PVS1 + PM2 + PP3)2c.1402 C > Tp.Arg468Maternal---D(0.913)D(1)D(37)D(1)--C(5.07)C (16.698) P (PVS1 + PS1 + PM2 + PP3)3c.1306_1309delp.Leu436Ilefs*15De novo----------- P (PVS1 + PS2 + PM2)4c.337G > Cp.Gly113ArgMaternal--D (0.001)D(0.990)D(1)D(27.8)D(1)-D(0.15)C(5.07)C(16.698)US(PM2 + PP3)5c.168G > Tp.Glu56AspMaternal--T(0.655)D(0.778)D(0.955)T (14.61)D(1)-D (0.285)C(2.91)NC (8.964)US(PM2 + PP3)Abbreviations: ACMG, american college of medical genetics and genomics; C, conserved; CADD, combined annotation-dependent depletion; D, damaging; EAS, east asian; FitCons, the fitness consequences of functional annotation; GERP, genomic evolutionary rate profiling; gnomAD, the genome aggregation database; LP, likely pathogenic; MAF, minor allele freuency; NC, non-conserved; P, polymorphism; PM2, absent in population databases; PP3, multiple lines of computational evidence support a deleterious effect on the gene/gene product; PS1, reported pathogenic variants (identical amino acid changes); PS2, de novo in a patient with the disease and no family history; PVS1, null variants (nonsense variants, frameshift variants, splicing variants in classical soil 1 or 2, start codon variants, single or multiple exon deletions) when the pathogenic mechanism of a disease is loss of function (LOF); SIFT, sorts intolerant from tolerant; T, tolerable; US, uncertain significanceNumber of algorithms predicted to be deleterious: total in silico algorithms, which was retrieved from the website http://varcards.biols.ac.cn/. Due to space limitations, only nine typical results were indicated in this table
The two missense variants were also not present in the gnomAD-all populations dataset. Additionally, at least five in-silico tools predicted these variants to be “damaging” (Table 2).
The five cases did not have any other pathogenic or likely pathogenic variants in genes known to be associated with seizure disorders [25].
Locations and molecular effect of SLC9A6 variants
The length of amino acid of SLC9A6 is 669. The NHE6 protein is a twelve-membrane spanning motif with the Na+/H + exchange occurring between transmembrane segments M4 and M5. There is a large and non-conservative cytoplasmic region at amino acid position 503-669 [26]. The variant c.337G > C/p.G113R, located at poor region, was associated with refractory epilepsy and speech delay, whereas the variant c.168G > T/p.E56D, which was located at loop region (Fig. 2A), was associated with pure epilepsy, suggesting a potential sub-molecular effect.
Fig. 2. Schematic illustration of variants location, hydrogen bond changes, and protein stability prediction. (A) Schematic diagram of SLC9A6 protein and the location of variants and their corresponding phenotypes. (B) Hydrogen bond changes and DDG values of SLC9A6 missense variants. The red dotted line represented hydrogen bonds. Variants with hydrogen bond changes or decreased protein stability are highlighted in red
The molecular effect of the missense variants was predicted by protein modeling using PyMOL 2.6 (Fig. 2B). The variant Gly113Arg was predicted to change hydrogen bonds with nearby residues and decrease protein stability.
Clinical features of the patients
In the present study, five hemizygous SLC9A6 variants were detected in five unrelated males with epilepsy and/or developmental disorders. The detailed clinical information was summarized in Table 1. The representative EEGs and MRs are shown in Fig. 3.
Fig. 3. Representative interictal electroencephalography (EEG) and magnetic resonance imaging (MRI) of the cases with SLC9A6 variants. (A) EEG of case 1 at the age of 5-years-old showed generalized electrical status epilepticus during slow-wave sleep. (B) EEG of case 2 at the age of 1-year-old showed spike-slow wave in bilateral frontotemporal and a mild degree of diffuse discharges. (C) EEG of case 4 at the age of 8-year-old showed sharp waves in bilateral central, parietal, and temporal discharge. (D) Interictal EEG of case 5 at the age of 11-year-old showed sharp waves in the left temporal region. (E) MRI of case 1 at the age of 3-year-old showed atrophy of the bilateral frontal and temporal poles. (F) MRI of case 5 at the age of 15-year-old showed hippocampal sclerosis
Fig. 4. Genotype-phenotype correlation of SLC9A6 variants. (A) Patients with null variants presented higher percentages of brain atrophy than those with missense variants (p = 0.003). (B) Patients with null variants presented higher percentages of microcephaly than those with missense variants (P<0.0001). (C) Patients with null variants presented higher percentages of motor disorders than those with missense variants (P = 0.002). (D) Patients with null variants presented earlier onset ages of seizures than those with missense variants (P = 0.0426)
The three cases with truncation variants were diagnosed as CS [27]. They presented with generalized epilepsy, with an onset age of nine months, 1 year, and 1 year and 7 months, respectively. They showed severe neurological developmental disorders and are unable to speak and walk independently. The head circumference of 44 ~ 45 cm, suggests microcephaly. The MRIs of the three cases showed cerebellar atrophy.
The two cases with missense variants presented with partial seizures, with onset ages of 5 years and 6 years, respectively. They did not present signs of microcephaly, ID, or motor developmental disorders. Case 4 showed language developmental delay. Brain MRI of case 4 was normal, and case 5 showed hippocampal sclerosis.
Two patients (cases 1 and 2) with truncation variants responded poorly to several anti-seizure medications. Among the two cases with missense variants, one achieved seizure-free (case 5) and the other one (case 4) was reduced by more than 50%.
In summary, patients with truncation variants showed CS, whereas patients with missense variants presented with epilepsy with/without developmental disorders.
Genotype-phenotype correlation
The present study showed that SLC9A6 missense variants are potentially associated with milder phenotypes than truncation variants. To get a full view of SLC9A6-associated phenotypes, we reviewed all previously reported SLC9A6 variants and explored possible mechanisms underlying phenotype variations. Until Nov 11, 2023, including the variants identified in the present study, a total of 55 variants, including 47 null and eight missense variants, were identified in 56 cases with detailed clinical data. The detailed clinical information of previously reported patients was summarized in Table S1.
The eight missense variants were identified in nine cases. The patients presented with a variety of phenotypes, ranging from partial epilepsy with favorable outcome to severe CS. Brain atrophy, microcephaly, and motor disorders were observed in 12.5% (1/8), 37.5% (3/8), and 62.5% (5/8), respectively, in the patients with missense variants. These manifestations, which were observed in 72% (26/36), 100% (45/45), and 100% (46/46), respectively, were generally more severe and uniform in patients with null variants. The proportions of these manifestations were significantly higher in patients with null variants than that of patients with missense variants (p = 0.0031, <0.0001, and 0.0023, respectively) (Fig. 4A-C). In addition, the seizure onset age of patients with null variants was significantly earlier than that of patients with missense variants (p = 0.0426) (Fig. 4D).
Sub-molecular effect
Given that damaging effects of variants are associated with severity of phenotype [28], we also analyzed the locations and molecular effects of previously reported SLC9A6 missense variants. Three variants were associated with CS, among which two (p.Pro157Leu and p.Gly186Arg) were located at the pore region and one (p.Gly351Asp) was located at the transmembrane region. They were predicted to change hydrogen bonds with nearby residues and decrease protein stability. The variant p.Arg170Gly, which was associated with early-onset epilepsy, was located at the loop region and was predicted to decrease protein stability. The other two variants, p.Glu579Lys and p.Leu584Phe, which were identified in patients with epilepsy with mental retardation or speech delay, respectively, were located at the c-terminal domain, among which one (p.Glu579Lys) was predicted to decrease protein stability (Fig. 2B). These findings suggested a sub-molecular effect, i.e., variants located within the pore or transmembrane region were associated with more severe damaging, thus resulting in more severe phenotypes.
Discussion
In the present study, we identified five SLC9A6 hemizygous variants in five unrelated males. All the variants did not present in the gnomAD- all populations and the missense variants were predicted to be damaging by multiple in silico tools. The three patients carrying null variants presented with refractory epilepsies and severe developmental delay; one patient harboring missense variant in the transmembrane region showed refractory epilepsies and speech delay; and one patient harboring missense variant located in the loop region achieved seizure with favorable outcome, suggesting a sub-molecular correlation. The results suggested that SLC9A6 variants are potentially associated with partial epilepsy with favorable outcomes.
The SLC9A6 gene encodes a sodium-hydrogen exchanger that is mainly expressed in the brain [26, 3]. Variants in SLC9A6 are associated with CS, a syndromic form of X-linked cognitive disability characterized by epilepsy, moderate to severe ID, progressive microcephaly, motor delay, and very limited language development [29–33]. Females with SLC9A6 variants may be less severely affected [1, 10, 34–36]. Previously identified variants were mainly null variants that resulted in protein termination. Including the variants identified in this study, a total of 47 null and eight missense variants are reported so far. Further analysis suggested that the proportions of brain atrophy, microcephaly, and movement disorders in patients with missense variants are significantly lower than that of patients with null variants. Patients with missense variants presented with significantly lower seizure onset age, tended to have milder intellectual impairments, and showed a better response to medications [37, 38] suggesting a genotype-phenotype correlation.
It was noted that the seven cases with missense variants exhibited a variety of phenotypes. The SLC9A6 gene encoded NHE6, which has 12 transmembrane helices and a regulatory C-terminal domain. There is a sodium-hydrogen exchanger (pore) region between transmembrane four and five [12]. Variants in the pore region or the transmembrane region were associated with CS, variants in the C-terminal resulted in epilepsy with mental retardation or speech delay, whereas variants in the loop region was associated with pure epilepsy, suggesting a sub-regional effect. Variants in the pore region or the transmembrane region were predicted to alter hydrogen bond with nearby residues and decreased protein stability, thus resulting in more severe phenotype.
The pLi and LOEUF of SCL9A6 is 0.998 and 0.206, respectively, suggesting that SLC9A6 is highly intolerant to loss of function variants. In animals, homozygous knock-out mice showed postnatal lethality. Previously reported variants are mainly null variants that result in severe neurodevelopmental delay [39–41]. The present study showed that SLC9A6 missense variants are potentially associated with mild epilepsy without developmental delay. Therefore, the phenotype spectrum of SLC9A6 potentially ranges from mild epilepsy with favorable outcome, to refractory epilepsy with neurodevelopmental disorders, even to early death, which is less often clinically diagnosed.
The SLC9A6 encodes a sodium-hydrogen exchanger that is a member of the solute carrier (SLC) family, a group of membrane transport proteins comprising over 300 members. In the brain, SLC transporters are involved in the transport of various solutes across membranes, suggesting that dysfunction of SLC transporters may contribute to epilepsy and neurodevelopmental disorders. Genes like SLC1A2, SLC2A1, SLC6A1, SLC12A5, SLC13A5, SLC25A22,* SLC35A2*, and SLC38A3 were previously found to be causative of developmental and epileptic encephalopathy [42–44]. SLC genes are also potentially associated with mild epilepsy. A typical example was SLC2A1, variants of which could cause late-onset epilepsy [45–48]. To our knowledge, this is the first report linking SLC9A6 variants to mild epilepsy with favorable outcome. Under natural selection pressure, genetic variants with less damaging effects would be more common than variants with more severe damaging effects, subsequently leading to milder and common diseases. Therefore, clinically, more attention should be paid to patients with mild epilepsies.
There are several limitations to this study. First, cases with missense variants are limited and more studies are needed to clarify the whole phenotype of patients with missense variants. Second, functional investigations of the detected variants are not performed.
Conclusion
Missense variants in SLC9A6 are potentially associated with partial epilepsy with favorable outcomes. The genotype-phenotype correlation and sub-molecular effects may be the underlying mechanisms of phenotypic variation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Padmanabha H, Saini AG, Sahu JK, Singhi P. Syndrome of X linked intellectual disability, epilepsy, progressive brain atrophy and large head associated with SLC 9A 6 mutation. BMJ case reports 2017, 2017.10.1136/bcr-2017-222050 PMC 578060129275387 · doi ↗ · pubmed ↗
- 2Zhang MW, Liang XY, Wang J, Gao LD, Liao HJ, He YH, Yi YH, He N, Liao WP. Epilepsy-associated genes: an update. Seizure; 2023. S 1059-1311(23)00254-6. & China Epilepsy Gene 1.0 Project 10.1016/j.seizure.2023.09.02137777370 · doi ↗ · pubmed ↗