Deciphering the near-complete genome and conducting pan-genome analysis of Brassica oleracea
Qiang Li, Yanhong Fu, Chenhao Zhang, Guoli Zhang, Yuqian Zhao, Ying Wang, Yajing Dou, Lin Gao, Shamsiah Abdullah, Xiao Ma, Yanbin Su, Xiaoming Song

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
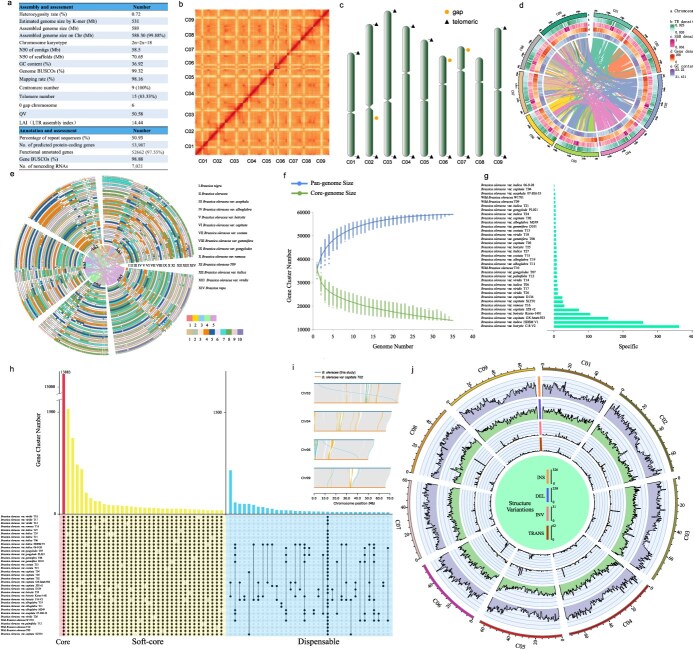 Figure 1
Figure 1- —Tangshan Normal University Research Project
- —Hebei Province Higher Education Institutions Science and Technology Research Project
- —S&T Program of Tangshan
- —National Natural Science Foundation of China10.13039/501100001809
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsChromosomal and Genetic Variations · Genomics and Phylogenetic Studies
Dear Editor,
Cabbage, scientifically known as Brassica oleracea, is a versatile vegetable from Brassica genus of Brassicaceae family. B. oleracea is highly valued for its nutritional, ornamental, and medicinal benefits. B. oleracea is classified into several morphotypes based on the form of the leaves and growth habit, such as B. oleracea var. capitata (head cabbage), B. oleracea var. acephala (kale), B. oleracea var. italica (broccoli), B. oleracea var. botrytis (cauliflower), B. oleracea var. gongylodes (kohlrabi), and B. oleracea var. gemmifera (Brussels sprouts).
The first genome sequencing of B. oleracea released in 2014 was a significant milestone in understanding the genetic makeup of Cabbage [1]. This research provided insights into the dynamics of Brassica genome evolution and served as an important resource for Brassica vegetable breeding. Recent advances in genomic sequencing have led to the assembly of high-quality genomes for different morphotypes of B. oleracea [1, 2]. However, there is still no near-complete genome for B. oleracea. Therefore, this study aims to resolve the first high-quality near-complete genome of B. oleracea, providing a higher quality genome for comparative and functional genomics research of B. oleracea. Moreover, based on this genome, all previously published B. oleracea genomes were collected and organized for pan-genome analysis.
To obtain the high-quality B. oleracea var. capitata genome, we performed the de novo genome sequencing using the latest sequencing technologies, including Oxford Nanopore Technology (ONT) ultra-long, PacBio HiFi, Illumina, and Hi-C technology. We selected a type of white cabbage as a representative B. oleracea for genomic sequencing research. First, the B. oleracea genome was estimated by K-mer using 26.45 Gb data from Illumina sequencing. The estimated genome size was 531 Mb, and the heterozygosity rate was 0.72% (Fig. 1a). The PacBio HiFi sequencer was adopted to generate 30.37 Gb (57.19 X) data with a mean length of 15.05 Kb.
Furthermore, Hi-C technology was used to anchor the assembled sequences to each chromosome, and a total of 54.93 Gb (103.45 X) data were obtained. The assembled genome length was 589.00 Mb, and the 588.30 Mb sequences anchored on the nine chromosomes with anchored ratio was 99.88% (Fig. 1a and b). The high-quality assembled genomes with the contig N50 was 58.50 Mb, and the scaffold N50 reaching 70.65 Mb (Fig. 1a).
Of particular importance is that ONT ultra-long sequences (27.00 Gb, 50.85 X) was used to achieve a near-complete level of genome. Six chromosomes (C01, C03, C04, C05, C08, and C09) have no gap, and the other three chromosomes (C02, C06, and C07) each have only one gap (Fig. 1a and c). This genome consists of nine chromosomes, with a total of 15 telomeres and 9 centromeres were detected. The read-mapping rate exceeds 98.16%, and the coverage is over 99.96% (Fig. 1a). The genome completeness is assessed by BUSCO as 99.32%, and the genome consistency quality value (QV) is 50.58. The genome LTR assembly index value is 14.44, which reached the high-quality reference level (Fig. 1a).
B. oleracea var. capitata has the most genome assemblies among its morphotypes, driven by its economic importance as a major crop and its status as the first sequenced subspecies in the B. oleracea [1]. However, although there are many versions of the genome, none of them have reached near-complete genome level. In this study, the genome of B. oleracea var. capitata was deciphered at the near-complete genome level. Compared to the genomes previously released, a higher quality and more complete B. oleracea genome was obtained. For example, our contig N50 was over 58.5 Mb, which was far larger than the previous report that the best contig is 31.3 Mb. Therefore, this study provides a more accurate and complete genome for the study of gene function and molecular breeding of B. oleracea.
Repetitive sequences constituted 50.93% of the B. oleracea genome. Specifically, Class I elements (retroelements) accounted for 25.96%, while Class II elements (DNA transposons) represented 24.97% (Fig. 1a and d). A total of 53 987 genes was predicted in B. oleracea genome, and 98.88% of BUSCO genes (1614) were detected, indicating high completeness of gene prediction. Among all the predicted genes, over 52 662 (92.16%) genes were annotated by GO, KEGG, KOG, Pfam, SWISS-PROT, TrEMBL, eggNOG, and NR databases. In regard to previous databases, 7021 noncoding RNA were found in B. oleracea genome (Fig. 1a).
In addition to our sequenced genome of B. oleracea var. capitata, we also collected genomes from 11 morphotypes of B. oleracea, B. rapa, B. nigra, and Arabidopsis thaliana to explore the evolutionary history of multiple B. oleracea morphotypes and representative Brassicaceae species.
Initially, by observing the homologous gene dot plots between 11 representative B. oleracea morphotypes and the reference species A. thaliana, we can infer the polyploidization events experienced by the studied species. A. thaliana has a relatively clear history of ancient polyploidization events, having undergone not only the whole-genome triplication (WGT) event (γ) that affected most angiosperms but also two additional whole-genome duplication events [1–4]. By examining the homology alignment between the genomes of B. oleracea morphotypes and A. thaliana, we can observe a clear 3:1 homologous in the gene dot plots between B. oleracea morphotypes and A. thaliana. This phenomenon also indicates that in addition to sharing ancient polyploidization with A. thaliana, the studied B. oleracea species have undergone an additional WGT event [1, 5]. Ultimately, a global alignment of the syntenic regions of the studied species is conducted using A. thaliana as a reference, and the overall display is presented in the form of a synteny circle map (Fig. 1e).
Based on our B. oleracea genome, and other collected 34 genomes from 11 B. oleracea morphotypes, we identified 59 086 nonredundant gene clusters, including 1 829 338 genes, which was used for constructing pan genome of B. oleracea (Fig. 1f–h). Here, the core cluster was defined as a cluster shared by all 35 genomes. There were 13 883 core clusters containing 723 739 genes (Fig. 1h). Among them, 20 873 genes in our genome are defined as core genes, 17 804 genes have been annotated with Gene Ontology (GO) functions. The function enrichment analysis showed that 12 GO terms were significantly enriched (p.adjust value <.05), such as pectin catabolic process, fatty acid biosynthetic process, auxin-activated signaling pathway, and photosynthesis. We further define the clusters present in 34 and 33 genomes as soft-core clusters. Among them, there were 9999 clusters, with a total of 443 960 genes. The 33 997 clusters (57.54%) present in 2–32 genomes contain 655 815 genes (35.85%) and were defined as dispensable clusters. The 1207 clusters existing solely in one genome contain 5824 genes and were defined as specific clusters (Fig. 1g). Among them, 614 genes in our genome are defined as specific genes and 431 genes have been annotated with GO functions. Notably, the GO functional enrichment analysis identified two significantly enriched terms, including nucleic acid binding (GO:0003676) and DNA integration (GO:0015074). However, the inconsistency of different genome versions annotated using different approaches may introduce some technical biases in the classification of gene clusters. Therefore, these specific classifications warrant more rigorous validation through more comprehensive pan-genomic investigations in future studies.
The comparative genome visualization map between our genome and that of B. oleracea var. capitata T02 reveals distinct improvements in certain regions of our genome. For instance, on chromosome 9, from about 32.5 to 40 Mb, significant enhancements are evident (Fig. 1i). Of course, such regions may also be due to structural variations between the two genomes. In this region, compared to our genome, B. oleracea var. capitata T02 has undergone 86 deletions, involving 91 genes, of which 87 genes have been annotated with GO functions. Furthermore, the nucleic acid binding and DNA integration terms were also identified by GO functional enrichment analysis. The KEGG annotation for these genes was H+-transporting ATPase (K01535). Furthermore, by aligning 34 B. oleracea genomes with our genome as reference, SVs (>50 bp) were classified into four categories, including insertions, deletions, inversions, and translocations. All nonrepetitive SVs include 155 581 insertions, 119 679 deletions, 1022 inversions, and 2289 translocations (Fig. 1j). We found that insertions and deletions are more likely to occur at both ends of chromosomes, while inversions and translocations are randomly distributed on nine chromosomes (Fig. 1j). These structural variations provide a rich data resource for better studying the morphological and genetic diversity of B. oleracea.
In conclusion, we present the first high-quality near-complete genome of B. oleracea and performed the large-scale pan-genome analysis. This genome and pan-genome analysis provided us with a wealth of data resources for functional genomics studies and molecular breeding of B. oleracea or even other Brassicaceae species.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Liu S, Liu Y, Yang X. et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Commun. 2014;5:393024852848 10.1038/ncomms 4930 PMC 4279128 · doi ↗ · pubmed ↗
- 2Guo N, Wang S, Wang T. et al. A graph-based pan-genome of Brassica oleracea provides new insights into its domestication and morphotype diversification. Plant Commun. 2024;5:10079138168637 10.1016/j.xplc.2023.100791 PMC 10873912 · doi ↗ · pubmed ↗
- 3Liu Z, Li N, Yu T. et al. The Brassicaceae genome resource (TBGR): a comprehensive genome platform for Brassicaceae plants. Plant Physiol. 2022;190:226–3735670735 10.1093/plphys/kiac 266PMC 9434321 · doi ↗ · pubmed ↗
- 4Feng S, Liu Z, Chen H. et al. PHGD: an integrative and user-friendly database for plant hormone-related genes. i Meta. 2024;3:e 16438868516 10.1002/imt 2.164PMC 10989150 · doi ↗ · pubmed ↗
- 5Wang X, Wang H, Wang J. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat Genet. 2011;43:1035–921873998 10.1038/ng.919 · doi ↗ · pubmed ↗