Expanding the Phenotypic Spectrum Associated with DPH5-Related Diphthamide Deficiency
Davide Politano, Cecilia Mancini, Massimiliano Celario, Francesca Clementina Radio, Fulvio D’Abrusco, Jessica Garau, Silvia Kalantari, Gaia Visani, Simone Carbonera, Simone Gana, Marco Ferilli, Luigi Chiriatti, Camilla Cappelletti, Katia Ellena, Elena Prodi, Renato Borgatti

TL;DR
This paper describes a new case of a rare genetic disorder caused by a mutation in the DPH5 gene, expanding the known range of symptoms associated with this condition.
Contribution
The study expands the phenotypic spectrum of DPH5-related diphthamide deficiency by reporting a milder variant with genotype–phenotype correlations.
Findings
The affected individual had a missense mutation (p.His260Arg) in DPH5, resulting in a milder phenotype compared to prior cases.
Clinical features included short stature, macrocephaly, heart defects, and developmental delays.
The phenotype overlaps with Noonan syndrome, suggesting differential diagnostic considerations.
Abstract
Background/Objectives: Neurodevelopmental disorders (NDDs) represent a clinically diverse group of conditions that affect brain development, often leading to varying degrees of functional impairment. Many NDDs, particularly syndromic forms, are caused by genetic mutations affecting critical cellular pathways. Ribosomopathies, a subgroup of NDDs, are linked to defects in ribosomal function, including those involving the synthesis of diphthamide, a post-translational modification of translation elongation factor 2 (eEF2). Loss-of-function (LoF) mutations in genes involved in diphthamide biosynthesis, such as DPH1, DPH2, and DPH5, result in developmental delay (DD), intellectual disability (ID), and multisystemic abnormalities. DPH5-related diphthamide deficiency syndrome has recently been reported as an ultrarare disorder linked to LoF mutations in DPH5, encoding a methyltransferase…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
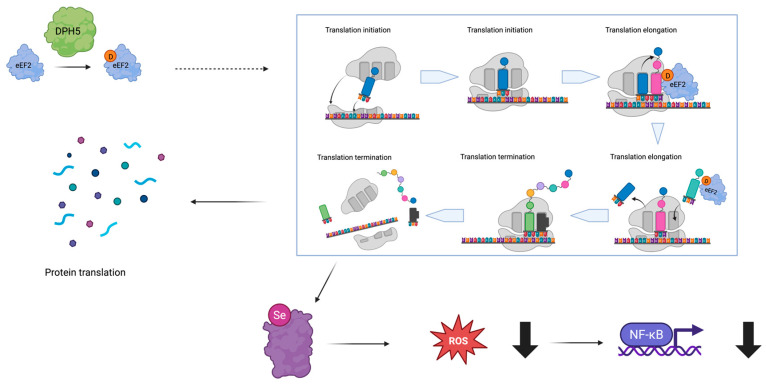 Figure 1
Figure 1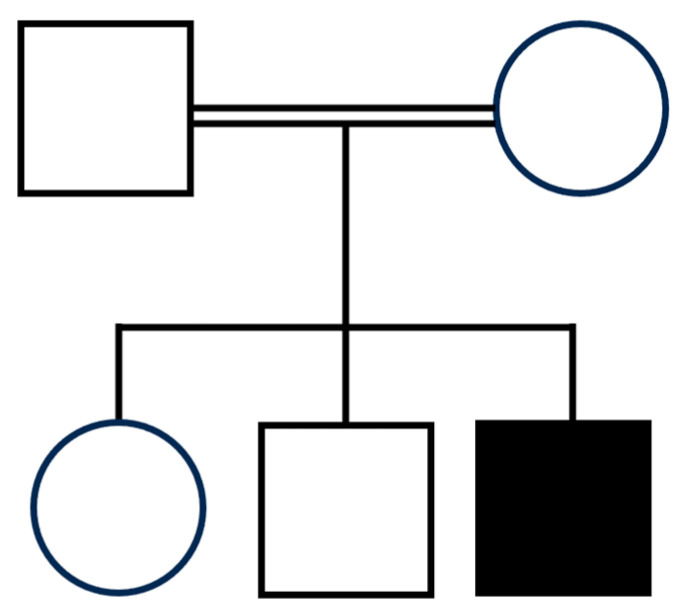 Figure 2
Figure 2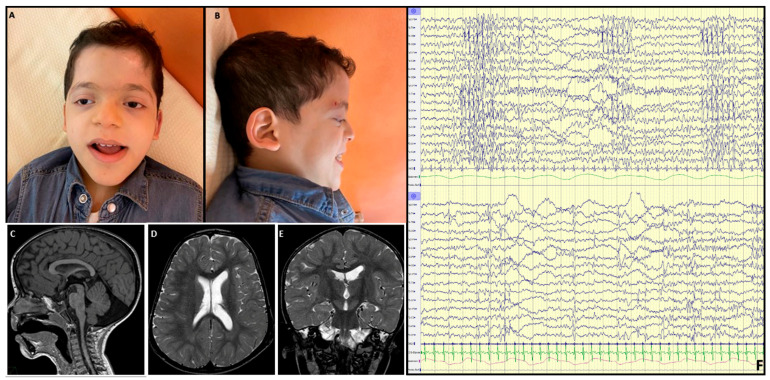 Figure 3
Figure 3- —Italian Ministry of Health
- —Ministry of University and Research (MUR)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPI3K/AKT/mTOR signaling in cancer · Interstitial Lung Diseases and Idiopathic Pulmonary Fibrosis · Neurogenetic and Muscular Disorders Research
1. Introduction
Neurodevelopmental disorders (NDDs) are a heterogeneous group of conditions affecting brain development and impairing the autonomy of affected individuals with variable severities [1]. NDDs may occur in the frame of complex syndromic entities characterized by multisystem involvement with organ malformations and/or extra-neurological functional alterations [2,3]. Up to 40% of isolated intellectual disability (ID) and syndromic NDDs have an established underlying monogenic defect, primarily due to de novo gene variants [4], and implicating various cellular pathways that are critical for brain development and function [5]. Among these disorders, ribosomopathies represent a relatively large group of clinically diverse and genetically heterogeneous diseases associated with altered ribosomal function due to defects involving ribosomal proteins, rRNA processing, or ribosome assembly [6,7].
Among ribosomopathies, deficiency in diphthamidation of eukaryotic translation elongation factor 2 (EEF2) due to biallelic LoF variants in genes encoding diphthamide synthesis enzymes is causally related to a phenotypic spectrum characterized by developmental delay (DD)/ID, short stature, craniofacial dysmorphisms, and ectodermal anomalies [8]. Diphthamide is a post-translationally modified histidine residue that is found in eukaryotic and archaeal translation elongation factor 2 (EF2), a protein with a crucial function in ribosome-dependent protein synthesis [9,10]. The biosynthesis of diphthamide is complex, involving multiple enzymes that are collectively known as diphthamide biosynthesis proteins (DPH), namely from DPH1 to DPH7, and the methylating cofactor S-adenosylmethionine (SAM) [11]. Alterations in the diphthamide biosynthesis pathway, specifically in DPH1 or DPH2 activity, have been associated with rare syndromic NDDs in humans [12,13,14].
In 2022, Shankar and colleagues provided compelling evidence causally linking LoF variants in the DPH5 gene, which encodes a key component of diphthamide synthesis, to a previously unrecognized NDD [15]. They described five patients from three unrelated families who shared a complex phenotype characterized by DD/ID, multisystemic abnormalities, and peculiar craniofacial dysmorphisms. Each family carried a unique set of biallelic variants in the gene, which were functionally validated to disrupt protein function.
DPH5, located on chromosome 1p21.2, is a highly conserved gene among eukaryotes. It is composed of eight exons and encodes different isoforms, which are the result of alternative transcript processing. DPH5 is a ubiquitously expressed protein that is part of a complex pathway required to synthesize diphthamide, a SAM-derived unique post-translational modification only found in archaeal and eukaryotic EF2 [9,10]. Diphthamide is specifically important in the regulation of selenoprotein translation and hence oxidative stress balance; thus, reduction in diphthamidation leads to NF-kb pathway hyperactivation (Figure 1) [16]. The NF-kb pathway is a very important and pleiotropic pathway in neuronal and non-neuronal cell development [17], strengthening the link between DPH5 activity alteration and syndromic NDDs. Diphthamide modification is dependent upon three complex series of reactions: the first biosynthetic step is carried out by the DPH1–DPH2 heterodimer that forms a 3-amino-3-carboxypropyl (ACP)-modified histidine upon electron donation by DPH3 and J-type chaperone DPH4; the second step leads to the generation of methylated diphthine from the ACP-modified intermediate by DPH5, followed by targeted demethylation reactions of conserved residues catalyzed by DPH7; the last step is the amidation reaction to generate fully diphthamide-modified eEF2 by DPH6 [11]. Consistently, LoF biallelic variants in three members of the above-mentioned associated pathway, specifically DPH1 [12], DPH2 [13,14], and DPH5 [15], have been associated with severe syndromic NDDs.
Here, we present the case of a 10-year-old boy affected by a multisystemic condition and carrying a homozygous pathogenic missense variant in DPH5, providing data that further expand the phenotypic spectrum associated with DPH5-related diphthamide syndrome.
2. Materials and Methods
Clinical, neurological, and dysmorphological evaluation was performed by a multidisciplinary team of clinical geneticists (F.S., S.G.), neuropsychiatrists (S.O., D.P., M.C.), and neuroradiologists (K.E., E.P.). Following signed informed consent from the patient’s parents, clinical data were acquired from the medical records in IRCCS Mondino Foundation (Pavia, Italy) and enrolled in the research project dedicated to undiagnosed patients, within the framework of Rete Italiana Salute dell’Età Evolutiva (Rete IDEA). The proband underwent a brain MRI 3T under sedation with acquisition of multiplanar T1- and T2-weighted images with age-appropriate TR and TE values.
To ameliorate the assessment of the craniofacial abnormalities, a computer-aided next-generation phenotyping tool named GestaltMatcher (University of Bonn, Bonn, Germany) was used [18]. GestaltMatcher is an extension of DeepGestalt and quantifies the facial syndromic similarity between two patients. It encodes each individual picture into a 320-dimensional facial phenotype descriptor (i.e., Clinical Face Phenotype Space—CFPS). A cosine distance was calculated in the CFPS to objectify the facial syndromic similarity between two pictures. A small distance between two pictures correlates to a high facial syndromic similarity. The pictures were ranked in ascending order based on the cosine distance. The facial phenotype of our patient was compared with the GestaltMatcher gallery.
Whole exome sequencing (WES) was performed on leukocyte-derived DNA samples using a trio-based approach. The SureSelect QXT Human All Exon V7 kit (Agilent, Santa Clara, CA, USA) was used for target region enrichment, and sequencing was conducted on an Illumina NovaSeq6000 platform (Illumina, San Diego, CA, USA). The raw data were processed using an in-house pipeline, as previously described [19,20], in line with the GATK’s Best Practices [21]. The UCSC GRCh37/hg19 genome assembly was used as a reference for read alignment, using the BWA-MEM tool [21,22], and variant calling was carried out using HaplotypeCaller (GATK v3.7, Broad Institute, Cambridge, MA, USA). SnpEff v.5.0 and dbNSFP v.4.2 tools were used for variants’ functional annotation [23]. Public (dbSNP150 and gnomAD V.2.0.1) and in-house (approximately 3300 exomes) databases were used to filter out variants with MAF higher than 0.001. Combined Annotation Dependent Depletion (CADD) v.1.64 [24], Mendelian Clinically Applicable Pathogenicity (M-CAP) v.1.0 [25], and Intervar v.2.0.1 [26] were considered for functional impact prediction. WES data output is summarized in Supplemental Table S1.
Sanger sequencing was carried out for confirmation of variants and segregation.
3. Results
3.1. Clinical Assessment
The proband was a 10-year-old boy, the third child born to non-consanguineous parents of Egyptian origin (Figure 2). Family history was negative for neuropsychiatric disorders. Pregnancy was regular, delivery occurred by cesarean section due to a previous cesarean section, and the perinatal course was depicted as normal except for moderate feeding problems that were characterized by chewing and swallowing difficulties. Motor development was severely delayed, with head and trunk control achieved at three years of age and independent, but unstable, walking at six years. Expressive language was absent. The patient experienced maintenance insomnia with frequent awakenings and episodes of apnea during sleep, starting in the first year of life. From 18 months of age, rare episodes of cyanosis, eye deviation, loss of consciousness, and hypertonia were observed, initially in the absence of a clear electroencephalographic consistency. At the age of seven, the frequency of these episodes increased, which were accompanied by electroencephalogram (EEG) abnormalities, including poor organization, and interhemispheric asymmetry with slower activity on the left hemisphere that was characterized by theta–delta rhythmic activity on left temporo-occipital region with spike and spike-and-wave epileptic anomalies that diffused contralaterally and worsened during sleep, in the absence of typical NREM sleep EEG features. Based on findings, pharmacological treatment was initiated, initially with valproic acid and clobazam, and later with valproic acid and levetiracetam, resulting in a relatively good seizure control.
At last evaluation (11 years and 4 months), the neurological and neuropsychiatric phenotype was characterized by profound ID with a happy disposition, absence of language, neuro-ophthalmologic abnormalities characterized by nystagmus, diffuse hypotonia, joint laxity, global muscle hypotrophy, pharmacologically responsive generalized epilepsy, and absent sphincter control. Craniofacial dysmorphisms were noted, including plagiocephaly, bilateral frontal and parietal bossing, features suggestive of a RASopathy (i.e., hypertelorism, bilateral mild ptosis, epicanthus, blue sclerae, coarse facial features, low set ears, depressed nasal bridge, and pointed chin), kyphosis, and pes planus (Figure 3 and Table 1). Multisystemic involvement was observed, including short stature, which was treated with growth hormone (GH) replacement therapy, frequent respiratory infections leading to multiple hospitalizations, moderate obstructive sleep apnea, and gastroesophageal reflux disease.
EEG documented a poor organization with multifocal spike and wave discharges, prevalent in temporo-posterior regions; polysomnography revealed moderate obstructive sleep apnea; brainstem auditory evoked potentials (BAEPs) analysis demonstrated a reproducible peak I, while peak III was not reproducible, and peak V was poorly reproducible. Brain magnetic resonance imaging (MRI) at the age of 3 showed callosal hypoplasia, multiple dilated perivascular spaces, a smaller left hippocampus with globular appearance, and mild inferior vermis hypoplasia associated with an enlarged cisterna magna (Figure 3C–F). Other instrumental examinations, including chest computed tomography, echocardiogram, and electrocardiogram–Holter, metabolic screening comprehensive of plasma and urinary amino acids and organic acids, and abdominal ultrasound, provided results within the normal limits.
3.2. Computational Facial Analysis
Due to the clinical impression of facial features suggestive of a RASopathy, we used the GestaltMatcher tool to objectify this finding [18]. Characteristic facial features were confirmed in the averaged facial analysis, showing YWHAZ-related disorder [27], Noonan syndrome (OMIM#163950) and cardiofaciocutaneous syndrome (CFCS, OMIM#115150) in the top 10 list with a distance of 0.657, 0.686, and 0.692, respectively. The same analysis was performed on subject 1B by Shankar et al. [15], who were homozygous for the same missense variant, returning Noonan syndrome and CFCS in the top 10 list with a distance of 0.631 and 0.702, respectively. While subjects 2A, 2B, and 3 show Noonan syndrome-like disorder with loose anagen hair (OMIM#607721), they show a distance of 0.733 and CFCS (distance 2B: 0.746 and 3:0.764) as the most similar RASopathies.
3.3. Molecular Analyses
Given the patient’s complex clinical presentation, genetic etiology was suspected, prompting genetic testing. Chromosomal microarray analysis revealed no pathogenic duplications or deletions. Trio-based WES analysis allowed for the identification of a homozygous missense variant (c.779A > G; p.His260Arg; NM_015958.3) in DPH5. This variant, previously reported in two siblings by Shankar et al. [15] and classified as pathogenic according to ACMG criteria (PM2, PS4, PS3, PP1, PP5), was found in gnomAD with a frequency of 0.000008. Segregation analysis confirmed the heterozygous state for the pathogenic variant in both parents.
4. Discussion
DPH5-related diphthamide deficiency syndrome is a recently identified and still poorly defined genetic disorder, with only five patients reported in the literature to date [15]. It is characterized by severe clinical manifestations, ranging from neonatal death to profound neurodevelopmental impairment, accompanied by craniofacial dysmorphisms, neurological and extra-neurological dysfunctions, and various malformations (see Table 1). It is included among ribosomopathies [21].
Ribosomopathies are genetic disorders caused by mutations in ribosomal constituents or in factors with a role in ribosome assembly, disrupting ribosome function and leading to tissue-specific clinical manifestations despite the universal need for ribosomes. Well-known examples include Diamond–Blackfan anemia, Shwachman–Diamond syndrome, dyskeratosis congenita, cartilage-hair hypoplasia, and Treacher Collins syndrome. The most prominent features include bone marrow failure, resulting in anemia and low blood cell counts, often diagnosed in early childhood, alongside developmental abnormalities such as growth retardation, craniofacial malformations, and limb defects. Some patients also display immune deficiencies, skin, nail, or hair changes, pancreatic or liver dysfunction, and NDDs. Notably, these conditions increase the risk of cancer, especially hematologic malignancies, with the risk more elevated for specific cancers. The disease course is often paradoxical, transitioning from hypo-proliferation (e.g., anemia) in early life to hyper-proliferation and cancer risk later—Dameshek’s riddle—with variable presentation and severity [28].
We herein describe the sixth patient affected by DPH5-related syndrome and the second family carrying the p.His260Arg variant. Our patient presented with a history of severe global DD, profound ID, nearly absent expressive language, nystagmus, craniofacial and body dysmorphisms, gastrointestinal issues, and growth retardation, as previously described in all the previously reported patients [15]. Additionally, he showed epilepsy, which had previously been reported in a subset of the originally identified affected individuals, as well as less common features, including sleep difficulties (40%), obstructive sleep apnea (40%), and frequent respiratory infections (40%). EEG findings were severely abnormal, and brain MRI showed marked, although non-specific, abnormalities across multiple brain regions consistent with the majority of patients previously reported [15]. Notably, BAEPs showed abnormalities in the reproducibility of waves originating from pontine structures, a novel finding not previously associated with this syndrome. Cardiac anomalies were not observed in our patient, although described in all the patients reported in the literature to date (see Table 1).
Furthermore, two additional patients (ClinVar accessions SCV003841335.1 and SCV004238160.1) carrying the same variant identified in our patient have been reported in ClinVar. The individual corresponding to SCV003841335.1 presented with seizures, hypogammaglobulinemia, malnutrition, and oropharyngeal dysphagia; no phenotypic information was provided for SCV004238160.1 (Variant information was retrieved from ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/, accessed on 25 June 2025).
The co-occurrence of short stature, which was responsive to GH supplementation therapy, relative macrocephaly, variable congenital heart defects, variable DD/ID, minor skeletal and ectodermal features, and recognizable craniofacial features (i.e., coarse facial features, bitemporal narrowing, bilateral frontal and parietal bossing, high forehead, epicanthal folds, hypertelorism, bilateral ptosis, deep-set eyes, broad and depressed nasal bridge, low-set ears, overfolded dysmorphic ears with thick earlobes, long deep philtrum, downturned corners of mouth, thick lips, pointed chin) suggests a differential diagnosis with Noonan syndrome and related conditions, as supported by GestaltMatcher analysis. Notably, these diseases, which are collectively termed RASopathies, are caused by germline pathogenic variants in genes that encode components and regulators of the RAS-mitogen-activated protein kinase (MAPK) signaling pathway [29,30,31]. DD/ID and speech delay/learning difficulties seem to be more severe in the DPH5-related syndrome, with seizures reported in a subset of individuals. Although a strict interaction between DPH5 and the RAS-MAPK pathway is not apparent, indicating the DPH5-related condition as a RASopathy “phenocopy”, Dph5 knockdown was demonstrated to ameliorate the pathogenic phenotypes in an in vitro model of Ras-induced tumor [32]. Moreover, the impact of activating mutations affecting the RAS-MAPK circuits results in NF-kb constitutive activation in several types of tumors [33,34], as expected in the dysregulation of selenoprotein translation [17] induced by DPH5-related diphthamide deficiency.
Shankar and colleagues generated a Dph5^His260Arg^ mouse model with a highly lethal phenotype in homozygous mice and a variably penetrant multisystemic disorder in heterozygote mice. The latter phenotype resembled the human biallelic phenotype, possibly due to the inbreeding and consequent susceptibility to other homozygous uncharacterized variants. They also confirmed, through in vitro phenotype assays using Saccharomices cerevisiae as a model system, that diphthamide synthesis was only partially compromised in strains expressing dph5His260Arg compared to the strains carrying nonsense and frameshift dph5 variants. Indeed, the latter types of variants determined a complete or severely defective protein function. In silico structural models performed to explore the consequences of the His260Arg substitution, although the residue being highly conserved accross species, did not document substantial conformational rearrangements of the protein or misfolding but showed a possible perturbation of the solvent-exposed surface of the protein binding to eEF2, predicting a reduced interaction between the two proteins [15].
Notwithstanding the limited available functional characterization of the variant, based on the relatively milder phenotype associated with the His260Arg substitution, we anticipate a likely hypomorphic behavior of this missense variant.
5. Conclusions
In conclusion, our case strengthens and broadens the clinical and radiological findings in DPH5-related syndrome and identifies His260 as a mutational hotspot associated with an overall milder phenotype, possibly resulting from residual DPH5 function.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Morris-Rosendahl D.J. Crocq M.-A. Neurodevelopmental disorders—The history and future of a diagnostic concept Dialogues Clin. Neurosci.202022657210.31887/DCNS.2020.22.1/macrocq 32699506 PMC 7365295 · doi ↗ · pubmed ↗
- 2Al Mutiri R. Malta M. Shevell M.I. Srour M. Evaluation of Individuals with Non-Syndromic Global Developmental Delay and Intellectual Disability Children 20231041410.3390/children 1003041436979972 PMC 10047567 · doi ↗ · pubmed ↗
- 3Fernandez B.A. Scherer S.W. Syndromic autism spectrum disorders: Moving from a clinically defined to a molecularly defined approach Dialogues Clin. Neurosci.20171935337110.31887/DCNS.2017.19.4/sscherer 29398931 PMC 5789213 · doi ↗ · pubmed ↗
- 4Brunet T. Jech R. Brugger M. Kovacs R. Alhaddad B. Leszinski G. Riedhammer K.M. Westphal D.S. Mahle I. Mayerhanser K. De novo variants in neurodevelopmental disorders-experiences from a tertiary care center Clin. Genet.2021100142810.1111/cge.1394633619735 · doi ↗ · pubmed ↗
- 5Parenti I. Rabaneda L.G. Schoen H. Novarino G. Neurodevelopmental Disorders: From Genetics to Functional Pathways Trends Neurosci.20204360862110.1016/j.tins.2020.05.00432507511 · doi ↗ · pubmed ↗
- 6Kampen K.R. Sulima S.O. Vereecke S. De Keersmaecker K. Hallmarks of ribosomopathies Nucleic Acids Res.2020481013102810.1093/nar/gkz 63731350888 PMC 7026650 · doi ↗ · pubmed ↗
- 7Nakhoul H. Ke J. Zhou X. Liao W. Zeng S.X. Lu H. Ribosomopathies: Mechanisms of Disease Clin. Med. Insights Blood Disord.2014771410.4137/CMBD.S 1695225512719 PMC 4251057 · doi ↗ · pubmed ↗
- 8Ütkür K. Mayer K. Khan M. Manivannan T. Schaffrath R. Brinkmann U. DPH 1 and DPH 2 variants that confer susceptibility to diphthamide deficiency syndrome in human cells and yeast models Dis. Model. Mech.202316 dmm 05020710.1242/dmm.05020737675463 PMC 10538292 · doi ↗ · pubmed ↗