Facile Diastereoselective Synthesis of Dihydroxyadipic Acid and Dihydroxyadipic Dilactone by Catalytic Reduction of Biosourced 3‑Hydroxy-2-Pyrone-6-Carboxylic Acid
Gabriella Leonardi, Aurora Bertuzzi, Ada Truscello, Cristian Gambarotti, Roberto Sebastiano

TL;DR
This paper presents a simple and selective method to synthesize dihydroxyadipic acid and its dilactone form from a renewable chemical source.
Contribution
The study introduces a novel catalytic reduction method for producing dihydroxyadipic acid and its dilactone with high diastereoselectivity.
Findings
Catalytic hydrogenation of 3-hydroxy-2-pyrone-6-carboxylic acid yields dihydroxyadipic acid with high diastereoselectivity.
The dilactone derivative was synthesized via intramolecular condensation and shows potential as a polymer comonomer.
A preliminary polymerization test with the dilactone and diamine was successfully demonstrated.
Abstract
3-Hydroxy-2-pyrone-6-carboxylic acid is a versatile chemical easily prepared from aldaric acids, a family of compounds obtained from renewable resources. Herein, an efficient synthesis of dihydroxyadipic acid by catalytic hydrogenation of this pyrone at room temperature and atmospheric pressure is reported. The reaction performed in organic solvents showed high diastereoselectivity, resulting in the possibility to obtain the racemic mixture of dihydroxyadipic acid (R,R) and (S,S) that was further converted to the dilactone 2,5-dioxabicyclo[2.2.2]octane-3,6-dione by a double intramolecular condensation reaction. This compound is a promising comonomer and/or chain-extender in the synthesis of polymers. As an example, a preliminary test of the polymerization reaction of the dilactone in the presence of a diamine is reported.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1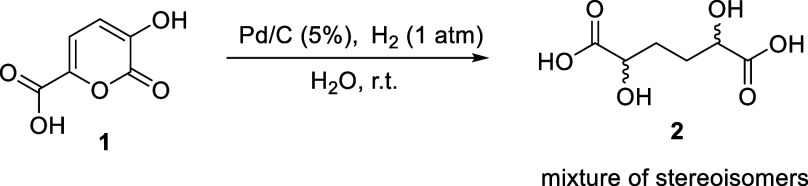 1
1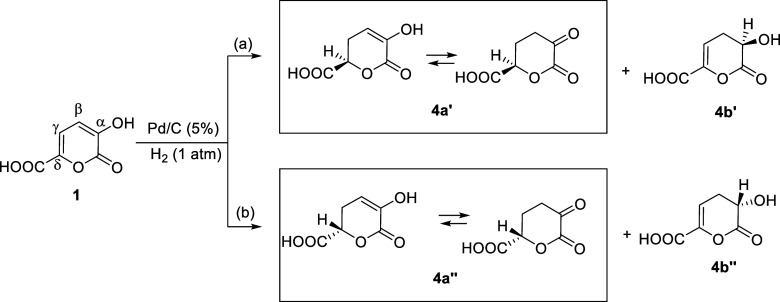 2
2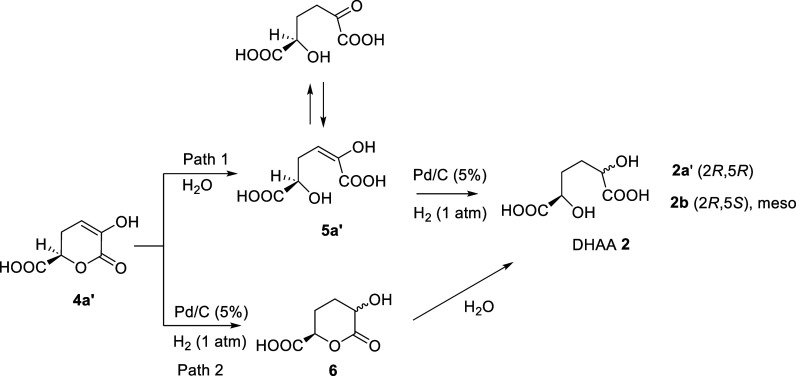 3
3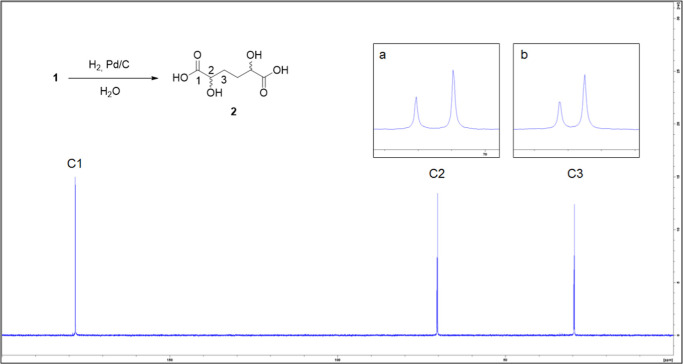 2
2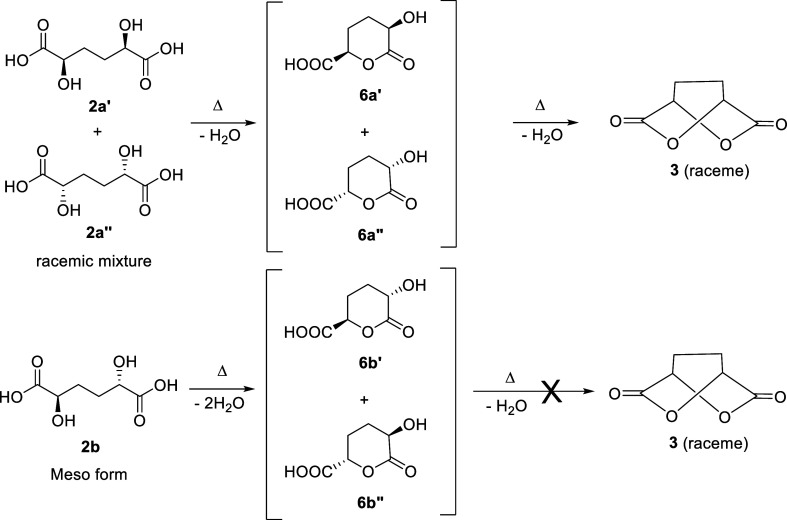 4
4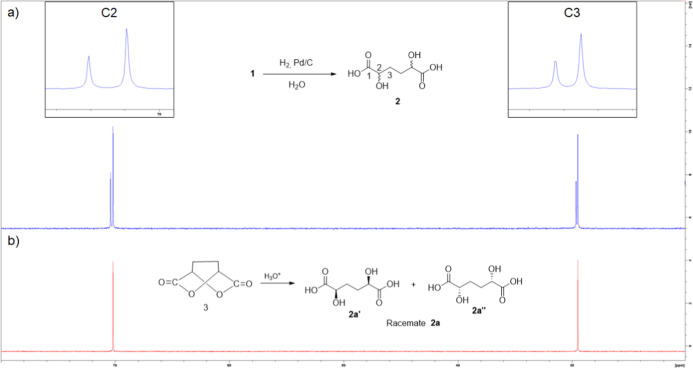 3
3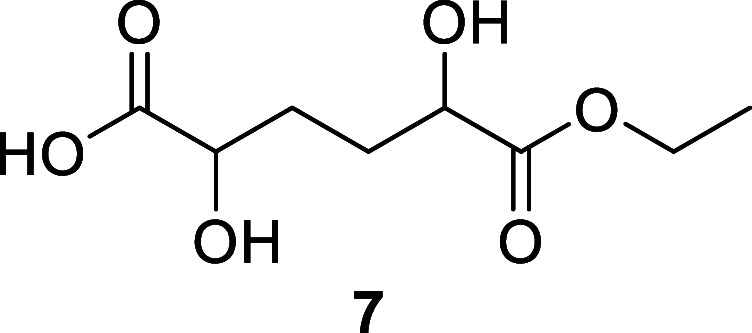 4
4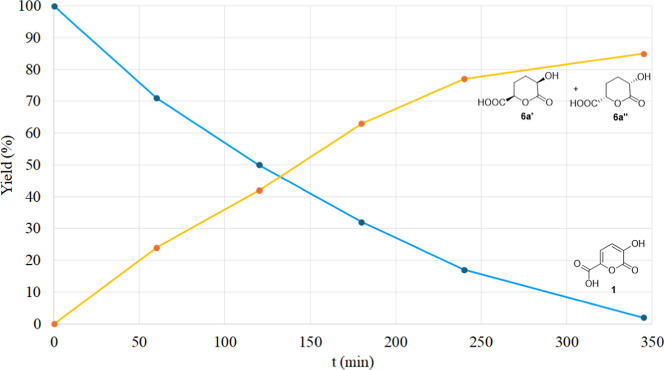 5
5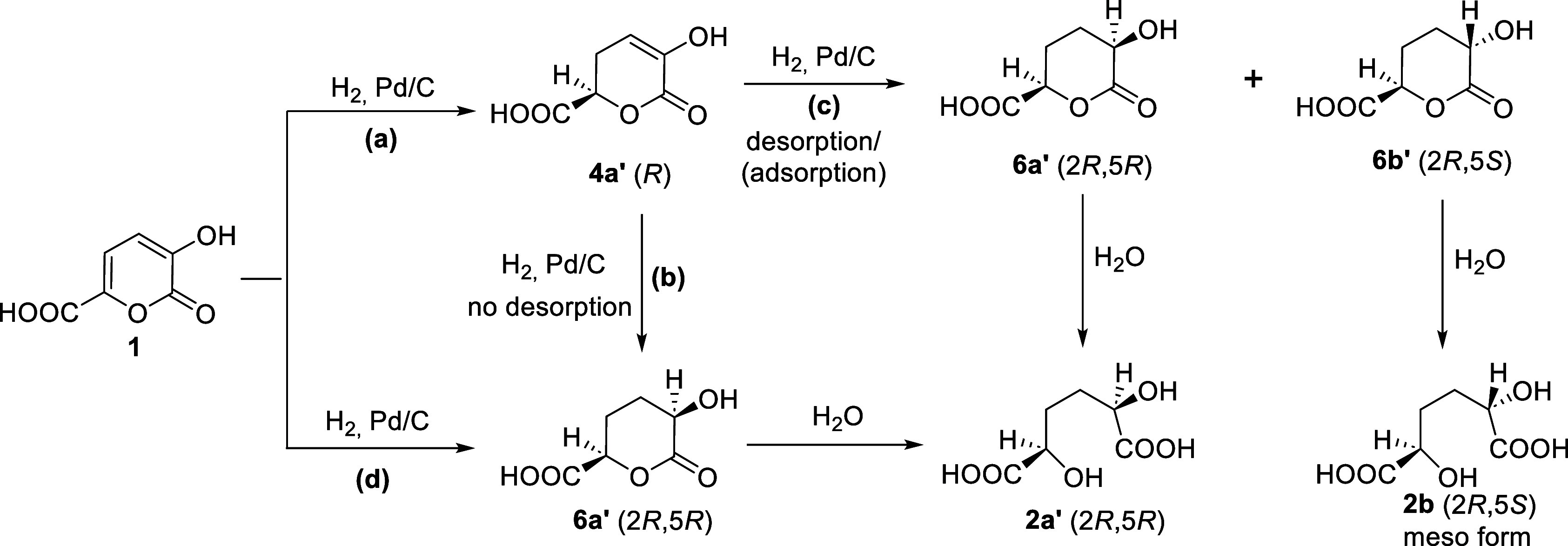 5
5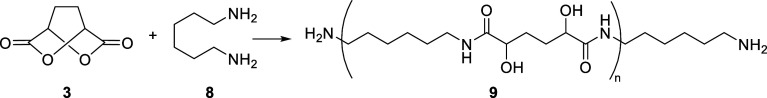 6
6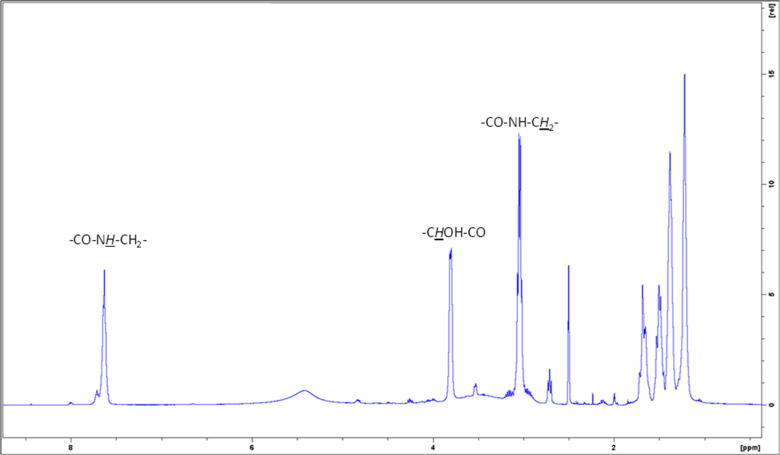 6
6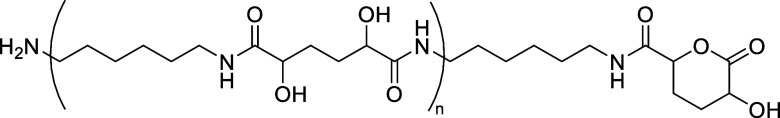 7
7Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCatalysis for Biomass Conversion · Enzyme Catalysis and Immobilization · Synthetic Organic Chemistry Methods
Introduction
The transition from fossil feedstock to renewable or waste sources in the preparation of organic compounds represents one of the most important goals in the field of sustainability. In particular, the synthesis of useful platform chemicals from agriculture waste biomass is one of the hot topics in scientific community. ?−? ? ? ?
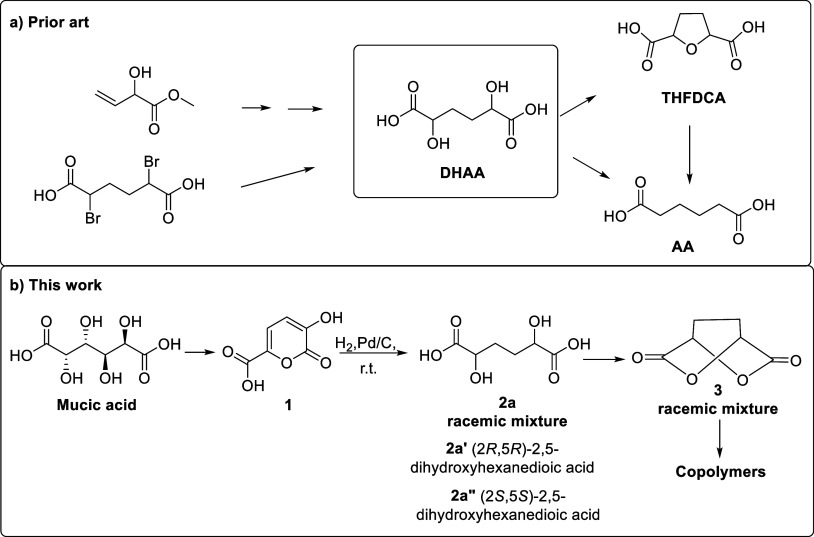
In the last years, several protocols have been developed for the preparation of adipic acid (AA), an important intermediate used in the production of nylons, from renewable resources using ecofriendly processes. ?−? ? ? ? ? ? ? ? ? Recently, aldaric acids, and their derivatives, have been studied as precursor of biobased AA. ?−? ? ? 2,5-Dihydroxyadipic acid (DHAA) was found to be an intermediate for the synthesis of tetrahydrofuran-2,5-dicarboxylic acid (THFDCA, Figurea) ?,? that in turn is a precursor of AA. ?,? Also, in a recent patent, DHAA itself is claimed to be a precursor of AA.?
Synthesis and use of DHAA: (a) prior art and (b) this work.
The use of DHAA is not limited to AA preparation as itself can be used as a monomer in the synthesis of polymers.?
The preparation of DHAA from adipic acid through the intermediate dibromo derivative was reported a century ago (Figurea).? The meso DHAA was also obtained, together with other coproducts, from gluconic acid in the presence of barium hydroxide at 140 °C.? Recently, DHAA has been prepared from sugar-derived methyl vinyl glycolate (Figurea),? by hydrogenation of aldaric acids over Pd/TiO_2_ performed at 2 MPa and 150 °C? and from 1,2,5,6-hexanetetrol in the presence of a platinum–bismuth.?
In recent years, our research group has been involved in the valorization of aldaric acids since these compounds are interesting polyfunctional molecules derived from polysaccharides present in biomass. In this context, we developed an efficient and ecofriendly synthesis of the pseudo aromatic 3-hydroxy-2-pyrone-6-carboxylic acid (3-hydroxy-2-oxo-2H-pyran-6-carboxylic acid, 1) from galactaric acid (mucic acid) (Figureb).? This compound has proven to be a versatile precursor in the synthesis of various aromatic compounds. ?,?
In this paper, the catalytic reduction under mild conditions of biosourced 3-hydroxy-pyrone 1 is reported (Figureb). The reaction performed in water led to a diastereoisomeric mixture of DHAA, whereas, in organic solvents, an almost complete diastereoselectivity was observed, obtaining, by hydrolysis, the racemic mixture (2R,5R and 2S,5S) of 2,5-dihydroxyhexanedioic acid (2a) (Figureb). Having raceme DHAA 2a the suitable configuration to allow the ring closure, it was possible to obtain the corresponding dilactone 2,5-dioxabicyclo[2.2.2]octane-3,6-dione (3) (Figureb). ?,? Dilactone 3, already used as a comonomer in the polymerization with lactide,? could be also used as a comonomer in the polymerization with diols or diamine to obtain biosourced polyhydroxyesters or polyhydroxyamides? and as a cross-linking agent. As an example, a preliminary test of polymerization of 3 with hexamethylenediamine is reported.
Results and Discussion
With the aim to perform the catalytic hydrogenation of hydroxypyrone 1 under mild and green conditions, we verified the possibility of carrying out the reaction in water at room temperature in the presence of hydrogen at atmospheric pressure and Pd/C as a catalyst. DHAA 2 was obtained in 90% yields within 18 h (Scheme). The structure was confirmed by comparison of ^1^H NMR spectrum with the data reported in literature.?
Hydrogenation of 1 in Water
The formation of DHAA 2 can proceed by following the steps reported in Schemes and ?. The first addition of hydrogen on pyrone 1 can occur from the “bottom” (Scheme, a) on the γ-δ double bond giving 4a′ and/or on α–β double bond giving 4b′ and respectively 4a’’ and/or 4b’’ from the “top” (Schemeb).
First Addition of Hydrogen on Pyrone 1 from the “Bottom” (a) and from the “Top” (b)
Possible Evolution of 4a′ to Dihydroxyadipic Acid 2 (See Scheme S1 for 4a’’)
As far as we know, in literature, it is generally reported that the first addition of hydrogen on pyrones occurs on the γ-δ double bond, ?−? ? but in those cases pyrone atoms in γ and δ are not substituted with groups that could give resonance effect with the double bond. For this reason, in our case, we cannot exclude the attack of hydrogen on the α–β double bond because of the presence of a carboxylic group.
Performing the reaction in water, the further hydrogenation of monounsaturated lactone 4a′ can proceed through two different pathways (Scheme). Following path 1, 4a′ initially undergoes hydrolysis to give the corresponding diacid 5a′, followed by hydrogenation obtaining the target DHAA as mixtures of diastereoisomers. In path 2, the second hydrogenation occurs before the hydrolysis of 4a′ forming the two diastereoisomer 6. The enantiomer 4a’’ would give the formation of the corresponding 2a” (2S,5S) and meso 2b (2S,5R) (Scheme S1). Similarly, monolactones 4b′ and 4b’’ can follow the same paths to give 2 (Scheme S2).
In principle, based only on the statistical distribution, the mixture should be composed by 50% of the raceme DHAA 2a (2a’ = 25%, 2a” = 25%) and 50% of meso DHAA 2b.
In literature, it is reported that hydrogenation of similar pyrones can be diastereoselective depending on the substrate and the reaction conditions. ?,? Therefore, an investigation of the stereoselectivity of the reaction was performed.
The quantitative ^13^C NMR analysis of the reaction mixture obtained by hydrogenation in water (Figure) showed a 1:2 ratio of the two DHAA diastereoisomers.
13C NMR (D2O) spectrum of 2 obtained by hydrogenation of 1. (a) Expansion of the region of C2; (b) expansion of the region of C3.
The identification the two diastereoisomers of DHAA 2 was performed by preparing the corresponding dilactone 3 by the thermal intramolecular condensation reaction (Scheme).? Dilactone 3 can be obtained only by the double lactonization of diastereoisomer 2a (racemic mixture of 2a′ and 2a”), via the formation of the intermediate monolactone 6a (racemic mixture of 6a′ and 6a”), as the OH and COOH substituents are in syn configuration. Conversely, the meso form 2b can afford only the monolactone 6b (racemic mixture of 6b′ and 6b”) (Scheme).
Lactonization of DHAA Diastereoisomers
Differently from that reported in the literature? where only a thermal treatment was used, we perform the lactonization by heating in a tinyclave the DHAA 2 dissolved in acetic acid. Dilactone 3 was then isolated by sublimation, analyzed by NMR, and identified by comparison with the data reported in literature.?
Dilactone 3 was then hydrolyzed in aqueous hydrochloric acid to form the corresponding DHAA 2a. By the ^13^C NMR spectrum of 2a (Figureb), it was possible to identify and assign the signals of the carbon bearing the hydroxyl group (C2) and the carbon of the methylene group (C3). Therefore, the signals present in the spectrum of the mixture of the two diastereoisomers 2 (Figures and ?a), obtained by hydrogenation of 1, can be correctly assigned.
13C NMR (D2O) spectra of (a) hydrogenation of pyrone 1 in water; (b) hydrolysis of dilactone 3.
This result indicates that the major product obtained in the catalytic reduction in water of pyrone 1 is represented by the racemic mixture of two enantiomers 2a′ and 2a’’.
It is known that the solvent can affect the selectivity of the hydrogenation of pyrones. ?,? In particular, as reported in Scheme, water can promote hydrolysis as well as the desorption from the catalyst. For these reasons, we investigated the reaction under the same conditions (room temperature and hydrogen at ambient pressure) in nonhydrolytic solvents such as acetic acid, THF, and ethanol.
Reactions were carried out up to no consumption of hydrogen, and the crude mixtures were analyzed by ^1^H NMR spectroscopy after removal of the solvent. Interestingly, the ^1^H NMR spectra of the reactions performed in THF or acetic acid showed the presence of a monolactone together with dilactone 3 and DHAA 2 (see Supporting Information, Figures S9 and 10). It has to be underlined that the ratios of these products, especially for experiments in acetic acid, depend upon the work up conditions as lactones are sensitive to temperature and moisture; for the same reason, all attempts in isolating monolactone failed. Thus, in an experiment performed in acetic acid, in which the catalyst was previously treated with acetic anhydride in order to remove moisture, two samples were withdrawn and the solvent was removed, respectively, at 70 °C under reduced pressure and at room temperature under a stream of nitrogen. The ^1^H NMR analyses (see Supporting Information, Figure S11) showed the monolactone as the major product in the sample treated at room temperature, whereas the dilactone was the major product present in the sample treated at 70 °C. Therefore, as the monolactone converted into 3 on heating, its structure was assigned to racemic monolactone 6a.
The reaction was performed in ethanol (Figure S12), resulted in an almost total conversion of 1 into the monoester 7 (Figure), and this latter was isolated and identified by NMR and MS spectroscopies.
Structure of monoester 7.
Being the DHAA 2 the target product, the mixtures of hydrogenations in THF, acetic acid, and ethanol, after the removal of catalyst and solvents, were submitted to hydrolysis. In Table, the overall isolated yields and the diastereoisomeric ratio 2 a /2b, determined by quantitative ^13^C NMR analysis, are reported.
1: Hydrogenation of Pyrone 1 : Isolated Yields of DHAA 2 (2a + 2b) and Diastereoisomeric Ratios 2 a /2b
Reactions performed in organic solvents (entries 2–4) showed, after hydrolysis, substantially the same yields and the same very good diastereoselectivity toward the racemic dihydroxyadipic acid 2a. The observed diastereoselectivity is comparable to that reported in literature for similar pyrones ?,? except in the case of alcoholic solvent, where no selectivity is reported.?
With the aim to monitor the hydrogenation during the time, the reaction in THF was also performed using a continuous H-Cube microhydrogenator apparatus, operating at 10 bar and 30 °C in the presence of 10% Pd/C. In this case, it was possible to analyze by ^1^H NMR spectroscopy the reaction mixture (see Supporting Information, Figure S14) just after removing the solvent at reduced pressure. The percentage of unconverted pyrone 1 and the analytical yields of the products were calculated by ^1^H NMR in the presence of an internal standard and are reported in Table S1 (see the Supporting Information). The corresponding data plots of 1 and 6a (racemic mixture of 6a′ and 6a’’) are drawn in Figure. Monolactone 6a is substantially the only product present in the reaction mixture. Only small amounts of DHAA 2 (1–2%) and dilactone 3 (1%) were identified in the ^1^H NMR spectra of the mixture, and their formation was probably due to the work-up procedure.
Hydrogenation of 1 performed in the H-Cube apparatus (H2 10 bar, 10% Pd/C, THF, 30 °C).
At low conversion, only very small signals, probably due to traces of dihydropyrone intermediates, were detected in the spectrum. Therefore, hydrogenation of these intermediates seems to be faster than that of the pyrone itself. As 6a was by far the major component after 345 min, the ^1^H NMR analysis allowed the assignment of the corresponding signals.
The diastereoselectivity 2a/2b observed (Table) can be justified considering the reaction mechanisms reported in Scheme. In order to simplify the discussion, only the enantiomers R (4a′) and R,R (6a′) are considered; their corresponding enantiomers 4a’’ and 6a’’ lead to the corresponding products (see the analogous Scheme S3 in the Supporting Information).
Catalytic Hydrogenation of 1: General Scheme
In the mechanism proposed, the adsorption/desorption processes of intermediate 4a′ from the catalyst surface are the major responsible of the diastereoselectivity. These processes depend on the nature of the solvent.
Regardless of the solvent used, the hydrogenation of 1 can occur following different paths: (i) formation of intermediate 4a′ followed by hydrogenation to give 6a′ without desorption from the catalyst surface (Schemea,b); (ii) formation of intermediate 4a′ and further hydrogenation to give 6a′ and 6b′ through desorption/adsorption from the catalyst surface (Schemea,c); and (iii) simultaneous hydrogenation of the two double bonds of 1 to give 6a’ (Schemed).
When water is used as the solvent, the hydrolysis of 4a′ to give 5a′ cannot be excluded as previously discussed (Scheme). Anyway 5a′ would evolve giving the same products through similar mechanisms.
Although the intermediate 4a′ has not been identified, its formation has to be considered as it allows us to justify the partial diastereoselectivity observed, in particular in the case of the reaction in water. In fact, the presence of the two diastereoisomers 2a and 2b can be explained only by assuming that part of 4a′ undergoes desorption from the catalyst and then adsorption on the other face of the ring. Thus, the further hydrogenation affords derivative 6b′ resulting in the observed loss of diastereoselectivity (Schemec).
As the hydrogenation of 1 performed in THF and in acetic acid (Table, entries 2,3 and Figure), after hydrolysis, showed a very high diastereoselectivity in 2a, the a, b, and d reaction pathways (Scheme) are the preferred ones. Conversely, when hydrogenation of 1 is carried out in water (Table, entry 1), low diastereoselectivity is observed (2a/2b ratio of 66:34), which indicates that the a and c reaction paths (Scheme) become competitive.
Similar to that discussed above, same products would be obtained also from 4b′ or 4b’’ (Scheme) and mechanism for 4b′ is proposed in the Supporting Information (Schemes S4).
As reported above, dilactone 3 was prepared in 12% yield starting from the mixture 66:34 of 2a/2b. Therefore, an attempt to maximize the yield of 3 was performed by running the dehydration of the reaction mixture containing almost pure 2a in a tinyclave at 140 °C using acetic acid as the solvent, obtaining 3 in 60% yield.
The dilactone 3 can represent a useful platform for the synthesis of copolymers as well as a branching agent. In this context, a very preliminary test of polymerization has been performed just to evaluate the possibility to use 3 as a comonomer. The reaction of dilactone with hexamethylenediamine 8 was performed at room temperature in DMSO-d 6 (Scheme).
Polymerization Reaction of 3 with Hexamethylenediamine 8
^1^H NMR analysis (Figure) after 24 h showed the presence of a mixture of products; the structure of the major component is compatible with the structure 9.
1H NMR spectrum (DMSO-d 6) of the polymerization mixture.
In the ^1^H NMR spectrum of 9 (Figure), the typical NH signal of the amide appears at 7.63 ppm, whereas at 3.80 ppm, there is a signal that could be assigned to the protons bound to the hydroxyl groups in the molecule. The methylene in α to NH of the amide appears at 3.04 ppm, whereas the small signals at 2.71 ppm could be assigned to the methylene protons bound to the nitrogen atom of the ending groups. The fact that the NH_2_ function has been found at both the end groups of structure 9 is suggested by the ESI-MS spectrum of the crude of polymerization reaction. The MS spectrum of the reaction mixture (Supporting Information, Figure S16) showed that the major part of the signals is that of 9 (n = 1–9), whereas only two signals seem to be ascribed to the presence of 10 (n = 1,2) (Figure). Anyway, in the ^1^H NMR spectrum, the signals compatible with the presence of 10 are very low.
Structure of the product with lactone as the end group 10.
The ^13^C NMR spectrum (Supporting Information, Figure S15) of the reaction mixture was recorded. The signal at 174.7 ppm confirmed the presence of the amide moiety.
In the ATR-FTIR (Supporting Information, Figure S17) of the solid precipitated from the reaction mixture, the peak at 1630 cm^–1^ can be ascribed to the amide CO stretching, whereas the peak at 1534 cm^–1^ is ascribed to the bending of the amide NH bond.
Conclusions
The very high diastereoselective synthesis of dihydroxyadipic acid starting from biosourced 3-hydroxy-2-pyrone-6-carboxylic acid 1 has been performed. The hydrogenation of 1 has been carried out in organic polar solvents in the presence of Pd/C under hydrogen at atmospheric pressure, at room temperature and without the need of asymmetric ligands, obtaining intermediate lactones that, after hydrolysis, led to the (R,R) and (S,S) racemic mixture of DHAA. The protocol showed a diastereoselectivity higher than 95%. A lower diastereoselectivity in the hydrogenation of 1 in water was found.
From the racemic mixture, it was possible to prepare the corresponding dilactone, a promising comonomer in the synthesis of polymers, and as an example, a preliminary test of the polymerization reaction in the presence of hexamethylenediamine is reported.
An attempt to discuss the very high diastereoselectivity observed is disclosed, and a mechanism which includes the possibility to have hydrogenation of the two double bonds of the pyrone without desorption from the catalyst is proposed.
Methods
A tinyclave BüchiglassBüchi AG (internal diameter of 2.5 and 10 cm length) was used for preparation of dilactone 3. A H-Cube Miniplus ThalesNano apparatus was used for the hydrogenation in THF. A Bruker AV 400 MHz instrument was used to record ^1^H NMR (400 MHz) and ^13^C NMR (100.6 MHz) spectra (100.6 MHz). ^1^H chemical shifts (δ) are given in parts per million relative to the residual proton of the solvent. ^13^C chemical shifts (δ) are given in ppm relative to the signal of the solvent; in the case of D_2_O, to the signals of DMSO (39.39 ppm) added to the sample as an internal reference. Quantitative ^13^C NMR spectra were acquired setting D1 = 20 s. MS analyses were performed with an Esquire 3000 plus ion-trap mass spectrometer equipped with an ESI source and with an Agilent 6420 Triple Quadrupole LC/MS system 6420 equipped with an ESI source. Melting points were determined with a Büchi 535 instrument and are uncorrected. ATR-FTIR spectrum was recorded using a Thermo Scientific Nicolet iS5 FT-IR Spectrometer with Nicolet id7 ATR unit (4000–400 cm^–1^ range). The collected data are analyzed with Thermo Scientific software OMNIC. Pyrone 1 was prepared from mucic acid by the procedure reported.?
Hydrogenation of 1 in Water
In a three-necked round-bottomed flask, 1.31 g of 3-hydroxy-2-pyrone-6-carboxylic acid (1) (purity 90%, 7.57 mmol) was dispersed in 26 mL of water. Then, 0.66 g of 5% Pd/C (50% moisture) was added to this suspension, and the resulting mixture was stirred at room temperature under the hydrogen atmosphere (ambient pressure) until no further hydrogen consumption was observed (18 h). The reaction mixture was then filtered, and the solvent was removed under reduced pressure to obtain 1.30 g (purity 94%, 90% yield) of 2 as a white solid. ^1^H NMR (DMSO-d 6): δ 3.98–3.88 (m, 2H), 1.78–1.65 (m, 2H),? 1.64–1.50 (m, 2H); ^1^H NMR (D_2_O, 4.79 ppm): δ 4.40–4.32 (m, 2H), 2.05–1.91 (m, 2H), 1.91–1.76 (m, 2H); ^13^C NMR (D_2_O + DMSO as reference 39.39 ppm): δ 178.1, 178.1, 70.4, 70.2, 29.7, 29.5.
Synthesis of Dilactone 3 by Thermal Dehydration
of 2
In a 25 mL tinyclave, DHAA 2 (obtained by hydrogenation in water) (0.512 g, purity 94%, 2.71 mmol) was dissolved in 15.0 mL of acetic acid. The solution was heated at 150 °C (oil bath) under stirring for 1 h and 50 min. After this time, the mixture was cooled to room temperature, and the solvent was removed under reduced pressure, obtaining 0.489 g of a white sticky solid. A sample of the solid was analyzed by ^1^H NMR spectroscopy in the presence of terephthalic acid as an internal standard. The analysis showed the presence of 3 in a 46% yield. The solid was then loaded in a sublimation apparatus and heated at 140 °C (oil bath), recovering 3 as a white solid (0.046 g, 0.324 mmol, yield 12%). M p 129–131 °C; ?,?
^1^H NMR (CDCl_3_): δ 5.01–4.99 (m, 2H), 2.44–2.33 (m, 2H), 2.25–2.13 (m, 2H);? ^1^H NMR (DMSO-d 6): δ 5.28–5.24 (m, 2H), 2.34–2.23 (m, 2H), 2.22–2.11 (m, 2H); ^13^C NMR (CDCl_3_): δ 166.6, 74.9, 23.2.
Hydrolysis of Dilactone 3 to Obtain 2a
Dilactone 3 (26 mg, 0.18 mmol) was dissolved in 2 mL of water and heated at 40 °C for 24 h. After this time, the solvent was removed, and 2a was obtained as a white solid. M p 135–136 °C. ^1^H NMR (D_2_O, 4.78 ppm): δ 4.40–4.34 (m, 2H), 2.05–1.94 (m, 2H), 1.89–1.76 (m, 2H). ^13^C NMR (D_2_O, spectrum of 2 used as reference): δ 178.2, 70.2, 29.5.
Hydrogenation of 1 in THF and Hydrolysis
In a round-bottomed flask, 0.525 g of 3-hydroxy-2-pyrone-6-carboxylic acid (1) (purity 96%, 3.22 mmol) was dispersed in 7 mL of THF. Then, 0.250 g of 5% Pd/C (50% moisture) was added to this suspension, and the resulting mixture was stirred at room temperature under the hydrogen atmosphere (ambient pressure) until no further hydrogen consumption was observed (44 h). The reaction mixture was then filtered, and the solvent was removed under reduced pressure. A sample of the residue was analyzed by ^1^H NMR spectroscopy in the presence of terephthalic acid as an internal standard. To the crude (0.598 g), 21 mL of water was then added, and the mixture was heated at 90 °C for 4 h. After this time, solvent was removed under reduced pressure obtaining 0.620 g (purity 90%, 3.1 mmol), 96% yield of 2 (2a/2b = 95/5).
Hydrogenation of 1 in Acetic Acid and Hydrolysis
In a round-bottomed flask, 0.516 g of 3-hydroxy-2-pyrone-6-carboxylic acid (1) (purity 96%, 3.16 mmol) was dispersed in 10 mL of acetic acid. Then, 0.253 g of 5% Pd/C (50% moisture) was added to this suspension and the resulting mixture was stirred at room temperature under the hydrogen atmosphere (ambient pressure) until no further hydrogen consumption was observed (25 h). The reaction mixture was then filtered, and the catalyst was washed 3 times with water. The aqueous solution recovered from the catalyst washing was combined with the acetic acid solution, and the solvents were removed under reduced pressure. A sample of the residue was analyzed by ^1^H NMR spectroscopy in the presence of terephthalic acid as an internal standard. To the crude, 20 mL of water was then added and the mixture was heated at 90 °C for 4 h. After this time, the solvent was removed obtaining 0.625 g (85% purity, 3.00 mmol), 95% yield of 2 (2a/2b = 96/4).
Hydrogenation of 1 in Acetic Acid
In a round-bottomed flask, 0.500 g of 5% Pd/C (50% moisture) was dispersed and stirred in 11 mL of a solution 10:1 of acetic acid and acetic anhydride for 1 h. After this time, the suspension was decanted, and the liquid was removed. In a three-necked round-bottomed flask, 1.01 g of 3-hydroxy-2-pyrone-6-carboxylic acid (1) (purity 98%, 6.33 mmol) was dispersed in 15 mL of acetic acid. The catalyst was then dispersed in 5 mL of acetic acid, and the resulting suspension was added to that of pyrone 1. Hydrogenation was carried out under stirring at room temperature (ambient pressure) until no further hydrogen consumption was observed. After 45 h, the mixture was filtered, two samples were withdrawn, and the solvent was removed, respectively, at 70 °C under reduced pressure and at room temperature under a stream of nitrogen. The samples were analyzed by ^1^H NMR spectroscopy.
Hydrogenation of 1 in Ethanol
In a round-bottomed flask, 0.281 g of 5% Pd/C (50% moisture) was washed, under stirring, with 5 mL of di EtOH, decanted, and then dispersed in 8 mL of ethanol. The resulting suspension was transferred in a round-bottomed flask containing 0.557 g of 3-hydroxy-2-pyrone-6-carboxylic acid (1) (purity 96%, 3.42 mmol). The resulting mixture was stirred at room temperature under the hydrogen atmosphere (ambient pressure) until no further hydrogen consumption was observed for 9 h. The reaction mixture was then filtered, and the solvent was removed under reduced pressure, obtaining 0.824 g of an oil. A sample of the residue was analyzed by ^1^H NMR spectroscopy in the presence of terephthalic acid as an internal standard. Part of this crude (0.103 g) was purified by flash chromatography on silica gel (AcOEt/AcOH 95:5 as eluent) obtaining 0.016 g of 6-ethoxy-2,5-dihydroxy-6-oxohexanoic acid 7 (0.078 mmol) as an oil.^1^H NMR (DMSO-d 6): δ 4.09 (dq, 2H, J = 7.1 and 1.0 Hz), 4.03–3.98 (m, 1H), 3.95–3.91 (m, 1H), 1.77–1.66 (m, 2H), 1.63–1.52 (m, 2H), 1.19 (t, 3H, J = 7.1 Hz); ^13^C NMR (DMSO-d 6): δ 175.6, 174.0, 69.4, 69.1, 59.9, 29.6, 29.5, 14.1; MS/MS (ESI): m/z 204.9 (15) [M – H]®, 158.9 (100) [M – EtOH-H]®.
Hydrogenation of 1 in Ethanol and Hydrolysis
In a round-bottomed flask, 2.06 g of 3-hydroxy-pyran-2-one-6-carboxylic acid (1) (purity 96%, 12.6 mmol) was dispersed in 40 mL of ethanol. Then, 1.01 g of 5% Pd/C (50% moisture) was added to this suspension, and the resulting mixture was stirred at room temperature under the hydrogen atmosphere (ambient pressure) for 24 h. The reaction mixture was then filtered, and the solvent was removed under reduced pressure. To the crude, 30 mL of water and 1 mL of trifluoroacetic acid were added and the mixture was heated at 90 °C for 5 h. After this time, the solvent was removed obtaining 2.76 g (74% purity, 11.4 mmol, presence of residual water as impurity), 90% yield of 2 (2a/2b = 96/4).
Hydrogenation of 1 in THF in H-Cube Apparatus
In a round-bottomed flask, 62.5 mg of 1 (purity 98% 0.393 mmol) was dissolved under stirring in 70 mL of THF and transferred in the H-Cube apparatus. The reaction was performed at 30 °C and at a pressure of 10 bar with a flow of the THF solution of 1 mL/min through a cartridge containing 1.2 cm^3^ of 10% Pd/C. Samples were withdrawn at the times as reported in Table S2, the solvent was removed and the residues were analyzed by ^1^H NMR spectroscopy using terephthalic acid as an internal standard. Results are reported in Table S2. ^1^H NMR (DMSO-d 6) of 6a: δ 4.97 (m, 1H), 4.22 (dd, 1H, J = 11.0 and 7.3), 2.25–2.05 (m, 2H), 2.00–1.90 (m, 1H), 1.65–1.52 (m, 1H).
Synthesis of Dilactone 3 by Thermal Dehydration
of a 95/5 Mixture of 2a/2b in Tinyclave
In a 25 mL tinyclave, a 93/7 mixture of 2a/2b (0.500 g, purity 77%, 2.15 mmol, obtained from the hydrolysis of the reaction mixture prepared by hydrogenation in THF) was dissolved in 20 mL of acetic acid. The reaction was heated in an oil bath set at 140 °C under stirring for 5 h. After this time, the mixture was cooled to room temperature and the solvent was removed under reduced pressure to obtain a white solid. The solid was transferred in a sublimation apparatus, heated in an oil bath at 140 °C at a pressure of 0.3 Torr recovering 3 as a white solid (0.173 g, purity 86%, 1.22 mmol, yield 60%.
Reaction of Dilactone 3 and Diamine 8
In a round-bottomed flask, 11.7 mg of dilactone 3 (purity 85%, 0.0700 mmol) and 9.23 mg of 1,6-hexamethylenediamine (8) (0.0794 mmol) were dissolved in 0.4 mL of DMSO-d 6 and the resulting mixture was stirred at room temperature for 24 h. After this, ^1^H NMR (Figure) and ^13^C NMR (Figure S15) spectra were recorded. ^1^H NMR (DMSO-d 6): δ 7.78–7.55 (m, 2H); 3.88–3.75 (m, 2H), 3.23–2.84 (m, 4H), 2.71 (t, 0.3, two end groups CH 2_NH_2), 1.75–1.58 (m, 2H), 1.57–1.44 (m, 2H), 1.44–1.31 (m, 4H), 1.28–1.14 (m, 4H); ^13^C NMR (DMSO-d 6): δ 174.7, 71.8, 39.0, 31.3, 30.1, 27.0 ppm. To the DMSO-d 6 solution, 0.4 mL of chloroform was added, obtaining a white solid. This latter was filtered, washed with chloroform, and ATR-FTIR was recorded (Figure S17). ATR-FTIR: ν 3387, 3295, 2924, 2855, 1630, and 1534 cm^–1^.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Blasi A.Verardi A.Lopresto C. G.Siciliano S.Sangiorgio P.Lignocellulosic Agricultural Waste Valorization to Obtain Valuable Products: An Overview Recycling 202386110.3390/recycling 8040061 · doi ↗
- 2Sheldon R. A.Green and sustainable manufacture of chemicals from biomass: state of the art Green Chem.20141695096310.1039/C 3GC 41935 E · doi ↗
- 3Bozell J. J.Petersen G. R.Technology development for the production of biobased products from biorefinery carbohydratesthe US Department of Energy’s “Top 10” revisited Green Chem.20101253955410.1039/b 922014 c · doi ↗
- 4Lin C. S. K.Pfaltzgraff L. A.Herrero-Davila L.Mubofu E. B.Abderrahim S.Clark J. H.Koutinas A. A.Kopsahelis N.Stamatelatou K.Dickson F.Thankappan S.Mohamed Z.Brocklesby R.Luque R.Food waste as a valuable resource for the production of chemicals, materials and fuels. Current situation and global perspective Energy Environ. Sci.2013642646410.1039/c 2ee 23440 h · doi ↗
- 5Sakuta R.Nakamura N.Production of Hexaric Acids from Biomass Int. J. Mol. Sci.201920366010.3390/ijms 2015366031357431 PMC 6695620 · doi ↗ · pubmed ↗
- 6Deng Y.Ma L.Mao Y.Biological production of adipic acid from renewable substrates: Current and future methods Biochem. Eng. J.2016105162610.1016/j.bej.2015.08.015 · doi ↗
- 7Corona A.Biddy M. J.Vardon D. R.Birkved M.Hauschild M. Z.Beckham G. T.Life cycle assessment of adipic acid production from lignin Green Chem.2018203857386610.1039/C 8GC 00868 J · doi ↗
- 8Unlu S.Niu W.Demirel Y.Bio-based adipic acid production: feasibility analysis using a multi-criteria decision matrix Biofuels, Bioprod. Bioref.20201479480710.1002/bbb.2106 · doi ↗