The m6A demethylase FTO regulates TNF-α expression in human macrophages following Toxoplasma gondii infection
Min Qin, Nan Gao, Jierui Sun, Shuqing Lin, Tingting Hu, Xinjian Liu, Rong Zhang, Yong Wang, Jingfan Qiu

TL;DR
This study shows how the m6A demethylase FTO helps regulate TNF-α expression in human macrophages infected with Toxoplasma gondii, revealing a new epigenetic mechanism.
Contribution
The study uncovers a novel post-transcriptional regulatory mechanism of TNF-α expression in human macrophages during T. gondii infection via m6A modification.
Findings
T. gondii infection increases FTO levels and decreases m6A modification in TNF-α mRNA.
Knocking down FTO dampens the immune response and allows uncontrolled parasite proliferation.
YTHDF2 binding to TNF-α mRNA is reduced, promoting TNF-α expression by inhibiting mRNA degradation.
Abstract
Toxoplasma gondii (T. gondii) is an opportunistic parasite. After infection, macrophages finely regulate the immune response to restrict parasite proliferation. It is well-known that N6-methyladenosine (m6A) plays a critical role in fine-tuning gene expression. To investigate whether m6A modification is involved in regulating the anti-infection immune response in human macrophages against T. gondii, this study utilized T. gondii tachyzoites from the RH strain to infect human THP-1 macrophages. qPCR and ELISA results show that T. gondii infection mounted the expression of TNF-α, IL-1β, and IL-6. Transcriptomic data suggest that the infection of T. gondii induced differential gene expression in pathways associated with TNF signaling and cytokine-cytokine receptor interaction. Meanwhile, expression of m6A regulators were evaluated using qPCR and Western blotting. T. gondii infection…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
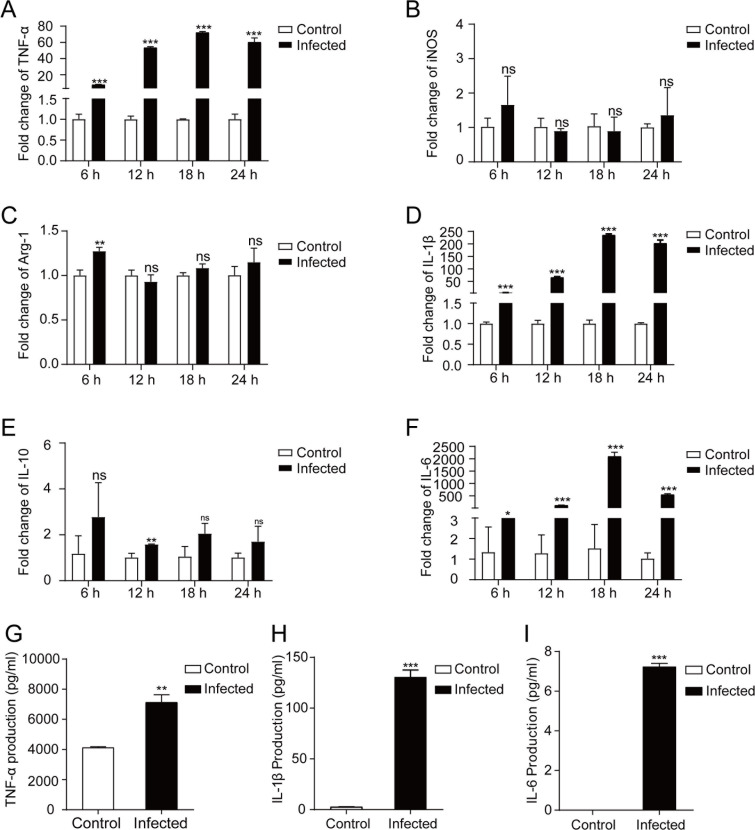 Fig 1
Fig 1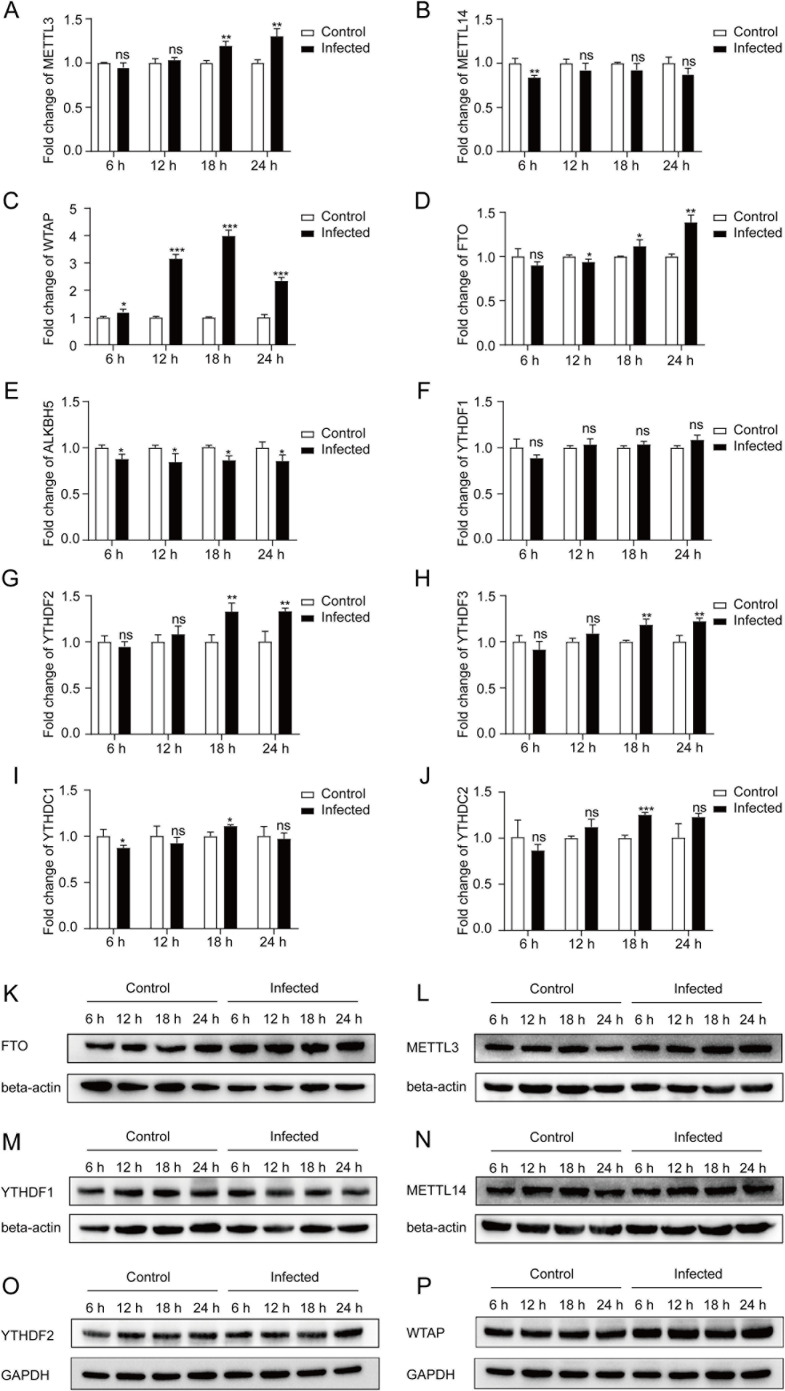 Fig 2
Fig 2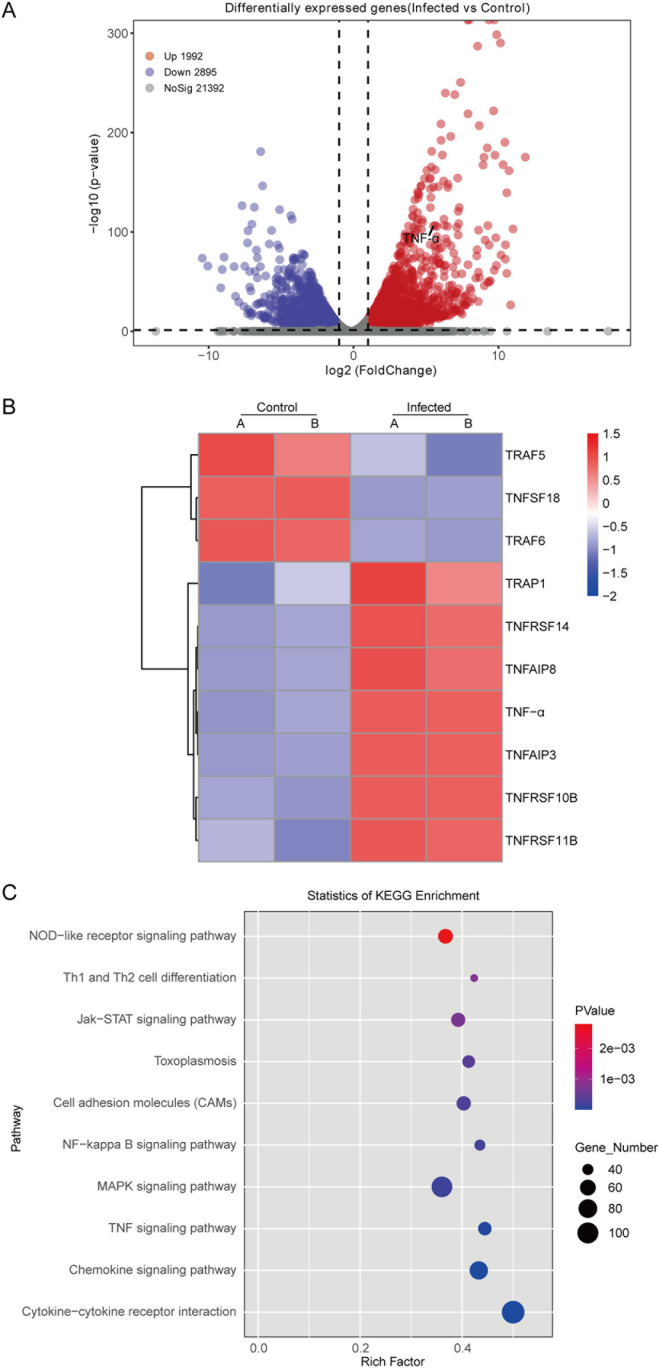 Fig 3
Fig 3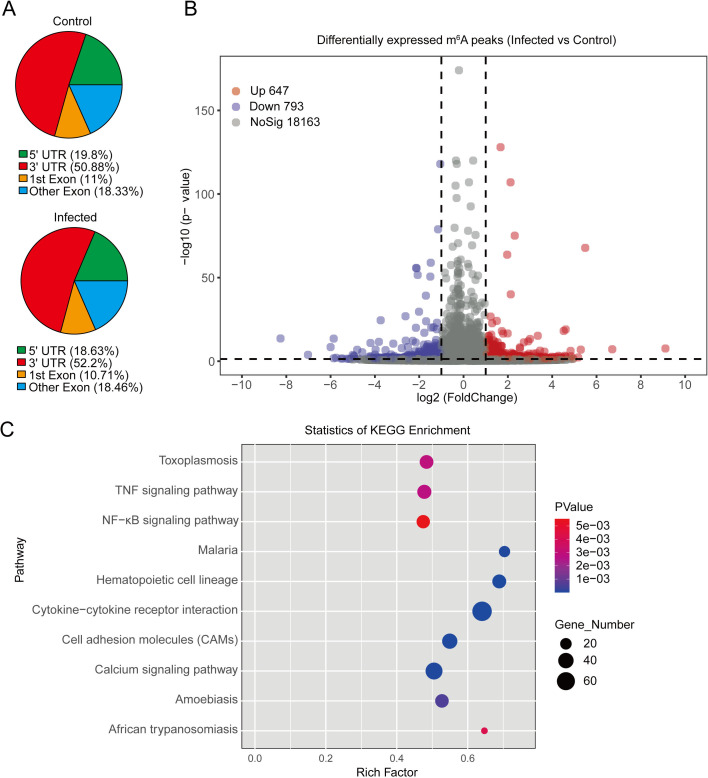 Fig 4
Fig 4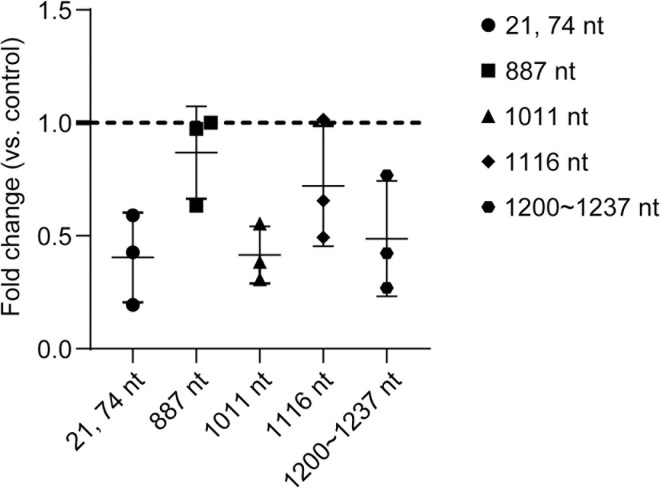 Fig 5
Fig 5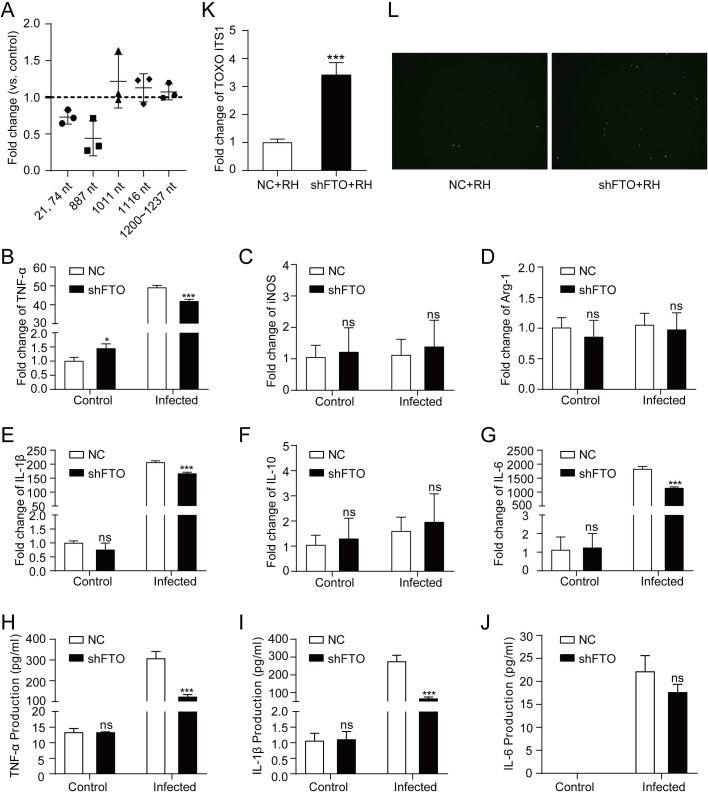 Fig 6
Fig 6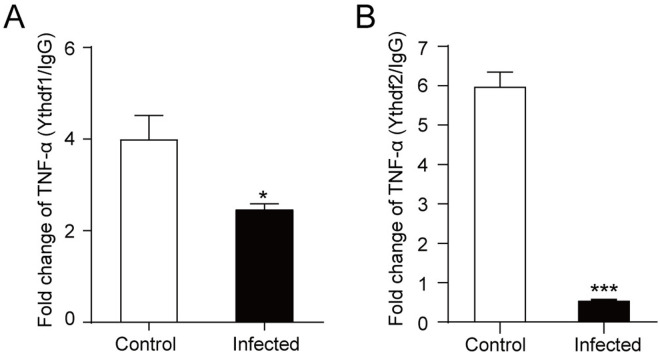 Fig 7
Fig 7- —http://dx.doi.org/10.13039/501100001809National Natural Science Foundation of China
- —http://dx.doi.org/10.13039/501100004608Natural Science Foundation of Jiangsu Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRNA modifications and cancer · HVDC Systems and Fault Protection · RNA Research and Splicing
Introduction
Toxoplasma gondii (T. gondii) is an important opportunistic parasite. In the healthy population*, T. gondii* infection does not cause obvious clinical symptoms [1,2]. However, infection occurs in the elderly and the immunocompromised individuals (such as AIDS patients, cancer patients and organ transplant recipients) could develop into acute toxoplasmosis, causing encephalitis, and even death [1,2]. Thus, the immune status of the host plays an important role in the development and outcome of T. gondii infection.
Macrophages play an indispensable role in resisting T. gondii infection and regulating immune homeostasis [3]. Among the inflammatory cytokines secreted by macrophages, tumor necrosis factor-α (TNF-α) has a highly pleiotropic effect in diverse cellular processes [4]. First of all, TNF-α is involved in controlling T. gondii infection. Blocking TNF-α may cause the latent infection of T. gondii to be reactivated, increasing the risk of acute toxoplasmosis [5]. Additionally, TNF-α could drive the conversion of tachyzoites to bradyzoites, resulting in the transition of toxoplasmosis from acute to chronic phase [6]. Furthermore, in spite of the protective role of TNF-α, excessive production of TNF-α can exacerbate inflammation and lead to immunopathology, contributing to the symptoms of acute toxoplasmosis, such as encephalitis [7]. Thus, it is of paramount importance to investigate the regulatory mechanisms of TNF-α after T. gondii infection.
In previous study, our research group focused on elucidating the transcriptional regulation mechanism of TNF-α. We demonstrated that MIC3 (microneme protein 3) activates TNF-α transcription via the TLR11/MyD88/NF-κB pathway in mouse macrophages [8]. In humans, T. gondii infection could also induce TNF-α production and human cells discriminate between viable and killed T. gondii tachyzoites [9–11]. However, human cells lack TLR11. Thus, we predicted that the mechanisms by which T. gondii regulates TNF-α expression in human macrophages are distinctly different from those in mouse macrophages.
It is well-known that post-transcriptional regulation helps cells respond more quickly to external stimuli than transcriptional regulation. Does post-transcriptional regulation play a role in modulating TNF-α expression in T. gondii-infected human macrophages? To answer this question, we focused on the most abundant internal mRNA modification, N^6^-methyladenosine (m^6^A), which has been reported as an important mechanism of post-transcriptional regulation [12]. m^6^A is an essential epitranscriptomic mark. Deposition of m^6^A is installed by a large protein complex called methyltransferases, consisting of METTL3, METTL14 and WTAP [13]. Demethylases, mainly FTO and ALKBH5, could remove existing m^6^A modification from mRNA [14,15]. The combination of methyltransferases and demethylases makes m^6^A modification a dynamic and reversible process. Finally, m^6^A modifications are directly recognized and bound by YTH-domain family proteins, including YTHDF1, YTHDF2, YTHDF3, YTHDC1 and YTHDC2 [16–18]. YTH-domain family proteins activate the downstream post-transcriptional regulation pathways, including promoting the degradation of mRNA.
In this study, we found that T. gondii infection significantly altered mRNA m^6^A modifications in human macrophages and increased the abundance of m^6^A demethylase FTO. Interestingly, there are m^6^A modification sites on human TNF-α mRNA. T. gondii could govern the expression of FTO to down-regulate m^6^A modification in the 3’UTR and 5’UTR regions of TNF-α mRNA, thereby mounting the TNF-α production in human macrophages. We believe that what we have learned about m^6^A and T. gondii-induced TNF-α production will provide important insights into the pathogenesis of toxoplasmosis and the T. gondii host-pathogen relationship.
Materials and methods
Ethics statement
This study was approved by the Ethics Review Board of Nanjing Medical University [permit no.: NJMU/ (2018) 498].
Study design
This study aimed to investigate the role of m^6^A modification in regulating the anti-infection immune response of human macrophages against T. gondii. Human THP-1 macrophages were infected with tachyzoites of T. gondii RH strain to establish an in vitro infection model. Uninfected cells served as controls. qPCR was performed to measure the mRNA abundance of inflammation-related genes. The secretion of inflammatory cytokines, including TNF-α, IL-1β, and IL-6, were measured by ELISA. The expression of m^6^A regulators was detected at the mRNA and protein levels using qPCR and Western blotting. RNA-seq was conducted to identify differentially expressed genes in T. gondii-infected macrophages compared to controls. m^6^A-seq was used to profile m^6^A modifications in mRNA transcripts. The m^6^A-IP-qPCR assay was performed to validate m^6^A modification levels in TNF mRNA. FTO was knocked down in THP-1 macrophages using lentiviral shRNA. Scramble shRNA served as the negative control. Infected FTO-knockdown cells were analyzed for m^6^A modification levels in TNF transcripts, expression of inflammation-related genes, secretion of inflammatory cytokines, and T. gondii proliferation. To investigate the role of m^6^A readers, RIP assay was performed to detect whether YTHDF1 or YTHDF2 had a direct effect on TNF-α mRNA.
Cell culture
THP-1 cells (human monocytic cell line) and HFF cells (human foreskin fibroblast cell line) were cultured in 1640 medium (Gibco BLR, MD, USA) and Dulbecco’s modified Eagle medium (Gibco BLR, MD, USA), respectively. Both media were supplemented with 10% fetal bovine serum (Gemini, CA, USA) and 1% 100 × penicillin and streptomycin (Gibco BLR, MD, USA). To induce differentiation into a macrophage phenotype, THP-1 cells were treated with 10 ng/ml phorbol myristate acetate (PMA, Sigma-Aldrich) for 72 h. For infection assay, THP-1 macrophages were infected with T. gondii RH tachyzoites and GFP-RH tachyzoites at a 1:1 ratio.
Preparation of T. gondii tachyzoites
T. gondii RH tachyzoites and GFP-expressing RH tachyzoites were serially subcultured in HFF cell lines. After observing 80% tachyzoite release, cell supernatants were filtered through a 3 μm pore size track-etched membrane (Shanghai Nengthink Filtration Technology Co. Ltd., Shanghai, China) to collect T. gondii RH tachyzoites and GFP-RH tachyzoites.
Real-time qRT-PCR
Total RNA was extracted from cell line samples and T. gondii tachyzoites using TRIZOL (Invitrogen, CA, USA). The quality and integrity of the extracted RNA for real-time qRT-PCR and m^6^A-IP-qPCR were evaluated using NanoDrop 2000 (NanoDrop, Wilmington, USA) and Bioanalyzer 2100 (Agilent, CA, USA). Samples with OD260/280 > 1.8, and RNA integrity number (RIN) > 7.0 were then reverse-transcribed to cDNA using HiScript III RT Supermix (Vazyme, Nanjing, China). Semi-quantitative RT-PCR, including cDNA, SYBR Green (Vazyme, Nanjing, China), and 10 μM primer pairs, was performed in a LightCycler 96 Instrument (Roche, Basel, Switzerland). Sequences of gene-specific primers used in this study are provided in S1 Table. Relative expression levels of these genes were calculated using the 2^−(△△Ct)^ method.
Enzyme-linked immunosorbent assay (ELISA)
After 24 h of T. gondii tachyzoites infection, THP-1 macrophage supernatants were harvested. The concentrations of TNF-α, IL-1β, and IL-6 in supernatants were detected using Human TNF-α, IL-1β, and IL-6 ELISA Kits (DaKeWe, Nanjing, China) respectively, following the manufacturer’s instructions.
Western blotting
For extracting total protein, THP-1 macrophages were lysed in RIPA lysis buffer (Millipore, MA, USA). After centrifugation at 12,000 g for 15 min at 4°C, the supernatants of lysates were collected. Protein concentrations were measured using a BCA assay (TIANGEN, Beijing, China). Protein samples were boiled with SDS-PAGE sample loading buffer (Beyotime, Shanghai, China) for 10 min. Appropriate amount of protein sample was resolved on a 10% SDS-PAGE gel, transferred to a 0.2 μm PVDF membrane (Millipore, MA, USA), and then blocked in TBST with 5% non-fat milk. Antibodies included N^6^-mA Methyltransferase (METTL3, METTL14, and WTAP) Antibody Sampler Kit (1:1000, Cell Signaling Technology), anti-FTO antibody (1:1000, Abcam), anti-YTHDF1 antibody (1:1000, Proteintech), anti-YTHDF2 antibody (1:1000, Proteintech), and HRP-linked anti-rabbit secondary antibody (1:2000, Cell Signaling Technology). Visualization was performed using ECL luminescence solution (FDbio Science, Hangzhou, China) on a ChemiDoc™ imaging system (Bio-RAD, CA, USA).
RNA-seq and m6A-seq
T. gondii RH strain tachyzoites were utilized to infect THP-1 macrophages, forming the infected group, while untreated THP-1 macrophages served as the control group. Total RNA was isolated using TRIzol reagent (Invitrogen, CA, USA). The quality and integrity of the extracted RNA were evaluated by NanoDrop 2000 (NanoDrop) and Bioanalyzer 2100 (Agilent), and were confirmed by agarose gel electrophoresis, as previously reported [19]. Samples with OD260/280 > 1.8, RIN > 7.0, and total amount > 50 μg were processed further. Ribosomal RNA of total RNA was depleted using Epicentre Ribo-Zero Gold Kit (Illumina, CA, USA). Magnesium RNA Fragmentation Module (NEB, MA, USA) was used to fragment RNA into small pieces for 7 min at 86°C. The fragmented RNA was saved for RNA-seq as input control. For m^6^A-seq, the cleaved RNA fragments and m^6^A-specific antibody (Synaptic Systems, Germany) were incubated for 2 h at 4°C in IP buffer. 50 mM Tris-HCl, 750 mM NaCl, and 0.5% Igepal CA-630 were used as IP buffer. Subsequently, the IP RNA was reverse-transcribed to create the cDNA, which were next used to synthesize U-labeled second-stranded DNAs. An A-base is then added to the blunt ends of each strand, preparing them for ligation to the indexed adapters. Then adapters are ligated to the fragments. After the heat-labile UDG enzyme (NEB) treatment of the U-labeled second-stranded DNAs, the ligated products are amplified with PCR by the following conditions: initial denaturation (1 cycle, 95°C for 3 min); amplification (8 cycles, 98°C for 15 s, 60°C for 15 s, and 72°C for 30 s); and then final extension (1 cycle, 72°C for 5 min). Finally, paired-end sequencing (PE150) was performed on an Illumina Novaseq™ 6000, and the data were analyzed by LC-Bio Technology CO., Ltd. (Hangzhou, China). All the sequencing experiments were performed with two biological replicates. The raw sequencing data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE288205. Raw reads obtained from the sequencing machine include reads containing adapters and low quality bases which will affect the following bioinformatics analysis. Thus, to get high quality clean reads, reads were further filtered by fastp (https://github.com/OpenGene/fastp, v0.19.4) [20]. For RNA-seq, genes with |log_2_Fold change| ≥ 1 and P < 0.05 were defined as differentially expressed genes (DEGs). DEGs were used to perform KEGG pathway enrichment analyses. For m^6^A-seq, DEGs with significantly altered m^6^A modification [false discovery rate (FDR) < 0.05] after T. gondii infection were used to perform KEGG pathway enrichment analyses.
m6A-IP qPCR
Firstly, total RNA was isolated using TRIZOL (Invitrogen, CA, USA) and its concentration was adjusted to 1 μg/μl. Subsequently, total RNA was fragmented for 5 min at 94°C in Veriti 96-Well Thermal Cycler (Thermo Fisher Scientific, MA, USA). 1 μg fragmented RNA was reserved as input control. The immunoprecipitation (IP) mixture was incubated for 2 h at 4°C, including m^6^A-specific antibody (Abcam) or mouse IgG (Beyotime), RnaseOUT (Invitrogen), Ribonucleoside vanadyl complexes (RVC, Sigma), and fragmented RNA. Meanwhile, Protein A/G Mix Magnetic Beads (Millipore, MA, USA) was blocked with BSA (Beyotime, Shanghai, China). Finally, magnetic beads were added into the incubated IP mixture for another 2 h at 4°C. The RNA in the magnetic beads was eluted twice and precipitated for further qRT-PCR detection.
FTO shRNA knock-down assay
Lentiviral shRNA was used to knock down of FTO in THP-1 cells. shRNA targeting sequences are shown below. Human FTO (shFTO): 5’-GGATGACTCTCATCTCGAAGG-3’. Scramble control sequence (NC): 5’-TTCTCCGAACGTGTCACGTAA-3’. Lentiviral particles harboring shRNA targeting FTO (shFTO) and negative control shRNA (NC) were designed and constructed by HANBIO (Shanghai, China). THP-1 cells were cultured in 6-well culture plates using Opti-MEM medium. Then cells were incubated in lentivirus-containing medium with polybrene (Sigma-Aldrich), and selected with puromycin (Solarbio, Beijing, China). The multiplicity of infection (MOI) of lentivirus was 60. FTO knock-down efficiency was assessed via qRT-PCR and Western blotting.
RNA-binding protein immunoprecipitation (RIP) assay
Anti-YTHDF1 antibody (Proteintech) or anti-YTHDF2 antibody (Proteintech) and Protein A/G Mix Magnetic Beads (Millipore) were pre-incubated overnight at 4°C. 20% percent of cell extracts were taken out as input control. Remaining cell lysates were incubated with previous magnetic bead-antibody complex for 4 h. After elution, RNA was isolated from IP fractions and input control for further detection.
Statistical analysis
Data from ELISA and qPCR assay was analyzed using Graphpad Prism software (GraphPad). Student’s t-test was used for comparisons between two groups. The P value < 0.05 was considered statistically significant.
Results
T. gondii infection upregulated inflammatory cytokine expression in human macrophages
During T. gondii invasion, T. gondii antigens interact with host immune cells to trigger the immune responses [21]. Therefore, to analyze the impact of T. gondii infection on human macrophages, we used T. gondii tachyzoites from RH strain to infect THP-1 macrophages. Inflammatory-related gene expression and the secretion of inflammatory cytokines were measured at 6 h, 12 h, 18 h and 24 h after infection (Fig 1).
*mRNA abundance of inflammatory-related genes and secretion of inflammatory cytokines in THP-1 macrophages infected with T. gondii tachyzoites.A-F: Transcriptional expression of inflammatory-related genes in THP-1 macrophages after T. gondii infection. mRNA abundance of TNF-α (A), iNOS (B), Arg-1 (C), IL-1β (D), IL-10 (E), and IL-6 (F) were quantitatively measured via qRT-PCR. Results are shown as fold changes compared with the control group. Data are represented as the mean ± SD (n = 3). Statistical significance was determined using Student’s t-test. ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001. G-I: Secretion of inflammatory cytokines in THP-1 macrophages after T. gondii infection. The secretion of TNF-α (G), IL-1β (H) and IL-6 (I) were detected by ELISA. Results are shown as fold changes compared with the control group. Data are represented as the mean ± SD (n = 3). Comparisons between two groups were made by Student’s t-test. **, P < 0.01; **, P < 0.001.
The mRNA abundance of TNF-α, IL-1β and IL-6 were significantly up-regulated from 6 h to 24 h post infection (Fig 1A, 1D, and 1F), while those of iNOS, Arg-1 and IL-10 did not change significantly from 18 h to 24 h post infection (Fig 1B, 1C, and 1E). The up-regulation amplitude was IL-6 > IL-1β > TNF-α, and the up-regulation amplitude increased with extended infection time within 18 h. Protein abundance of TNF-α, IL-1β, and IL-6 increased at 24 h after infection (Fig 1G, 1H, and 1I), consistent with mRNA abundance. Interestingly, unlike mRNA abundance, protein abundance of TNF-α exhibited the largest increase after T. gondii infection (Fig 1G). In sum, the results showed that T. gondii tachyzoites could induce the expression of inflammatory-related genes and promote the secretion of inflammatory cytokines, including TNF-α, IL-1β and IL-6.
T. gondii infection increased the expression of m6A methyltransferase WTAP and demethylase FTO
In the regulation of host immune response against pathogens, post-transcriptional regulation responds more rapidly than transcriptional regulation. RNA m^6^A modification plays an important role in post-transcriptional regulation in various pathogen infection models [22–25]. After T. gondii infection, the transcriptional expression of methyltransferases, demethylases and m^6^A-specific binding proteins (m^6^A reader proteins) were detected by qRT-PCR (Fig 2A to 2J). At different time points post-infection from 0 h to 24 h, WTAP was highly up-regulated at the transcriptional level, accompanied by a slight decrease in demethylase ALKBH5 (Fig 2C and 2E). Meanwhile, the m^6^A demethylase FTO also showed an upward trend from 18 h to 24 h after T. gondii infection (Fig 2D). The mRNA abundance of methyltransferase METTL3, and m^6^A-specific binding protein YTHDF2 and YTHDF3 significantly increased from 18 h to 24 h after infection (Fig 2A, 2G, and 2H).
*mRNA and protein abundance of m6A methyltransferases, demethylases and m6A-specific binding proteins in THP-1 macrophages infected with T. gondii tachyzoites.A-J: 6A methyltransferases, m6A demethylases and m6A-specific binding proteins in THP-1 macrophages after T. gondii infection. mRNA abundance of METTL3 (A), METTL14 (B), WTAP (C), FTO (D), ALKBH5 (E), YTHDF1 (F), YTHDF2 (G), YTHDF3 (H), YTHDC1 (I), and YTHDC2 (J) were quantitatively measured by qRT-PCR. Results are shown as fold changes compared with the control group. Data are represented as the mean ± SD (n = 3). Comparisons between two groups were made by Student’s t-test. ns, P > 0.05; *, P < 0.05; **, P < 0.01; **, P < 0.001. K-P: Western blotting analysis for m6A methyltransferases, m6A demethylases, and m6A-specific binding proteins. Protein abundance of FTO (K), METTL3 (L), YTHDF1 (M), METTL14 (N), YTHDF2 (O), and WTAP (P) were measured by Western blotting.
Then, the protein abundance of m^6^A methyltransferases, demethylases and m^6^A-specific binding proteins were measured at 6 h, 12 h, 18 h, and 24 h after infection. The results showed a significant increase in WTAP and FTO compared to the control group (Fig 2K and 2P). As for METTL3, METTL14, YTHDF1, and YTHDF2, T. gondii infection causes slight or no significant changes in protein abundance (Fig 2L-2O).
T. gondii infection affected TNF signaling in human macrophages
To thoroughly evaluate the effects of T. gondii infection on THP-1 macrophages, we performed global gene expression profiling using RNA-seq. |log_2_Fold change| ≥ 1 and P < 0.05 were used as criteria to identify DEGs (differentially expressed genes) between the infected group and the control group. After 24 h-infection of T. gondii in THP-1 macrophages, a total of 4887 DEGs were screened, including 1992 up-regulated genes and 2895 down-regulated genes (Fig 3A). Clustering analysis indicated a significant upregulation of TNF-α expression level after T. gondii infection (Fig 3B). Additionally, the expression of several members of the TNF super-family, including TRAP1, TNFAIP3, and TNFAIP8, were notably altered. KEGG pathway enrichment analysis showed that DEGs were enriched in TNF signaling pathway, toxoplasmosis pathway, cytokine-cytokine receptor interaction pathway, NF-κB signaling pathway, and so on (Fig 3C). These results suggest that T. gondii infection could induce an immune response in human macrophages. Proinflammatory cytokine TNF-α significantly changed after infection.
Significantly altered genes and pathways of macrophages after T. gondii infection.A: Volcano plot of differentially expressed genes. The red dots indicate up-regulated DEGs after T. gondii infection. The blue dots indicate down-regulated DEGs after T. gondii infection. The gray dots indicate genes without any significant change. Genes with |log2Fold change| ≥ 1 and P < 0.05 are defined as DEGs. B: Heatmap of DEGs related to TNF pathway after T. gondii infection. The blue color indicates down-regulated genes. The red color indicates up-regulated genes. C: KEGG pathway enrichment analysis of DEGs. The size of the dot is proportional to the number of enriched DEGs, and the rich factor indicates the degree of enrichment.
T. gondii infection influenced RNA m6A modification in human macrophages
To systematically analyze changes in RNA m^6^A modification in T. gondii-infected THP-1 macrophages, we performed m^6^A-seq. After 24 h-infection of T. gondii in THP-1 macrophages, m^6^A peaks were mainly distributed in the 3’ UTR and the 5’ UTR regions in both control and infected groups (Fig 4A). T. gondii infection increased methylation in the 3’ UTR region, while methylation in the 5’ UTR region decreased. Differentially methylated m^6^A peaks (|log_2_Fold change| ≥ 1 and P < 0.05) were identified, with 1440 total (647 up-regulated and 793 down-regulated) in the infected group compared to the control group (Fig 4B). KEGG pathway enrichment analysis of DEGs with significantly altered m^6^A modification (FDR < 0.05) after T. gondii infection revealed that such genes were enriched in toxoplasmosis pathway, TNF signaling pathway, NF-κB signaling pathway, and so on (Fig 4C).
Transcriptome-wide m6A methylation profiling of THP-1 macrophages by m6A-seq after T. gondii infection.A: Distribution of m6A peaks across the transcriptome of THP-1 macrophages. B: Volcano plot of differentially methylated m6A peaks. The red dots indicate significantly up-regulated m6A methylation after T. gondii infection (log2Fold change ≥ 1 and P < 0.05). The blue dots indicate significantly down-regulated m6A methylation after T. gondii infection (log2Fold change ≤ -1 and P < 0.05). The gray dots indicate m6A methylation without any significant changes. C: KEGG pathway enrichment analysis of DEGs with significantly altered m6A modification (FDR < 0.05) after T. gondii infection. The size of the dot is proportional to the number of enriched DEGs, and the rich factor indicates the degree of enrichment.
T. gondii infection downregulated the m6A modification in the 5’UTR and 3’UTR region of TNF-α mRNA in human macrophages
The m^6^A modification is not uniformly distributed in different mRNAs. Given the importance of TNF-α in responding to T. gondii infection, we probed the m^6^A modification sites on TNF-α mRNA. Firstly, the online prediction tool SRAMP was used to predict the potential m^6^A modification sites on human TNF-α mRNA. The results showed that there were 9 potential m^6^A modification sites in the 5’UTR and 3’UTR regions of TNF-α mRNA (S2 Table). The m^6^A-seq results suggested that m^6^A modifications in the 5’UTR and 3’UTR region of TNF-α mRNA decreased after T. gondii infection. Furthermore, we used m^6^A-IP-qPCR to verify the above findings. The m^6^A modification levels decreased at sites both in the 5’UTR region and in the 3’UTR region after T. gondii infection (Fig 5).
Detection of TNF-α mRNA m6 A sites after T. gondii infection.The m6A modification sites on TNF-α mRNA were detected by m6A-IP-qPCR. Fold change = 1 indicates no change (n = 3).
Knock-down of FTO attenuated TNF-α expression and led to uncontrolled proliferation of T. gondii in human macrophages
FTO, a key m^6^A demethylase, was upregulated in THP-1 macrophages after T. gondii infection. To investigate the function of FTO in T. gondii-induced immune response in macrophages, we knocked down FTO in THP-1 macrophages by shRNA (S1 Fig). In FTO knock-down cell lines, m^6^A modification levels at sites in the 5’UTR and 3’UTR regions of TNF-α mRNA did not decrease significantly after T. gondii infection (Fig 6A), while T. gondii infection down-regulated the m^6^A modification levels at sites in the 5’UTR and 3’UTR regions of TNF-α mRNA in wildtype cells (Fig 5). These results indicate that FTO plays a role in T. gondii-induced down-regulation of m^6^A modifications in TNF-α mRNA.
*Effects of FTO knock-down on THP-1 macrophage immune response induced by T. gondii.A: Changes of m6A modification on TNF-α mRNA in FTO knock-down cell lines after T. gondii infection. Results are expressed as fold change relative to the control group. Fold change = 1 indicates no change (n = 3). B-G: mRNA abundance of inflammatory-related genes, including TNF-α (B), iNOS (C), Arg-1 (D), IL-1β (E), IL-10 (F), and IL-6 (G), were quantitatively measured via qRT-PCR. Results were shown as fold changes compared with the control group. Data were expressed as the mean ± SD (n = 3). Comparisons between two groups were made by Student’s t-test. ns, P > 0.05; *, P < 0.05; ***, P < 0.001. H-J: Secretion of TNF-α (H), IL-1β (I), and IL-6 (J) were detected by ELISA. Results were shown as fold changes compared with the control group. Data were expressed as the mean ± SD (n = 3). Comparisons between two groups were made by Student’s t-test. ns, P > 0.05; **, P < 0.001. K: The mRNA abundance of TOXO ITS1 was measured by qRT-PCR. L: The proliferation of T. gondii GFP-expressing tachyzoites was observed by fluorescence microscope.
In the shFTO group, mRNA abundance of TNF-α, IL-1β, and IL-6 was significantly lower than that in the control group, while the transcriptional levels of iNOS, Arg-1, and IL-10 remained unchanged (Fig 6B-6G). The levels of secreted cytokines after T. gondii infection decreased significantly for TNF-α and IL-1β in the shFTO group compared to the control group, and the secretion of IL-6 in the shFTO group showed a decreasing trend but was not statistically significant (Fig 6H-6J). FTO knock-down weakened the immune response induced by T. gondii, as evidenced by decreased expression of inflammation-related genes and reduced secretion of inflammatory cytokines.
Additionally, after T. gondii infection, the mRNA levels of TOXO ITS1, which indicate parasite burden of T. gondii, increased significantly in the FTO knock-down group compared with the control group (Fig 6K). Meanwhile, enhanced proliferation of T. gondii green fluorescent protein (GFP)-expressing tachyzoites was also observed by the fluorescence microscope in the FTO knock-down group (Fig 6L). These results further demonstrated that FTO knock-down weakened the anti-infection immune response against T. gondii.
T. gondii infection inhibited YTHDF2 binding to TNF-α mRNA
YTHDF proteins are known to bind m^6^A sites and could promote the decay of m^6^A-modified mRNA [26]. An RNA-binding protein immunoprecipitation (RIP) assay was performed to detect whether YTHDF1 or YTHDF2 had a direct effect on TNF-α mRNA. After T. gondii infection, decreased binding to TNF-α mRNA was observed for YTHDF1 (about 2-fold, *P *< 0.05, Fig 7A). Notably, the YTHDF2 RIP assay showed that after T. gondii infection, the binding of YTHDF2 to TNF-α mRNA significantly down-regulated (about 11-fold, *P *< 0.001), indicating T. gondii infection remarkably weakened the binding of YTHDF2 to TNF-α mRNA (Fig 7B). This could be attributed to the reduced m^6^A modifications on TNF-α mRNA after T. gondii infection. These findings suggest that T. gondii infection may mount TNF-α expression by inhibiting the degradation of TNF-α mRNA.
*YTHDF1 RIP and YTHDF2 RIP.A: qRT-PCR detection of the changes of TNF-α enriched by YTHDF1 and IgG. B: qRT-PCR detection of the changes of TNF-α enriched by YTHDF2 and IgG. Results were shown as fold changes compared with the control group. Data were expressed as the mean ± SD (n = 3). Comparisons between two groups were made by Student’s t-test. *, P < 0.05; **, P < 0.001.
Discussion
This study focuses on the post-transcriptional regulation mediated by m^6^A modification in human macrophages after T. gondii infection, especially the role of m^6^A in post-transcriptional regulation of TNF-α and the effect of T. gondii infection on it. In this study, m^6^A-seq and RNA-seq were conducted to systematically observe the changes of RNA m^6^A modification after T. gondii infection. Our transcriptome analysis showed that the expression level of TNF-α was significantly up-regulated in T. gondii-treated THP-1 macrophages. Further KEGG pathway analysis indicated that DEGs were enriched in TNF signaling pathway and cytokine-cytokine receptor interaction pathway. Based on the m^6^A-seq data, the m^6^A peaks were mainly enriched in the 3’UTR and the 5’UTR regions. In the infected group, the m^6^A methylation in the 3’UTR region was increased, concomitant with decreased methylation in the 5’UTR region, suggesting that the mRNA splicing, stability and translation were affected. In addition, KEGG pathway enrichment analysis of DEGs with significantly altered m^6^A modification showed that DEGs in toxoplasmosis pathway, TNF signaling pathway, NF-κB signaling pathway were highly enriched. All these findings suggest that there is a close relationship between m^6^A modification alterations and T. gondii-induced immune responses, especially the pro-inflammatory cytokine TNF-α.
TNF-α biosynthesis is regulated by a variety of complex mechanisms, which mainly occur in gene transcription, mRNA transport, stability and translation, intracellular signal transduction and other stages [27–30]. At the transcriptional level, TNF-α expression is co-regulated by transcription factors, co-regulators, and chromatin modifications [27]. Our research group found that MIC3, which is a T. gondii excretory-secretory antigen, could induce the immune response of mouse macrophages through TLR11/MyD88/NF-κB signaling pathway, and initiate the TNF-α production against T. gondii. Post-transcriptional regulation is also essential in TNF-α synthesis. Different microRNAs (miRNAs) and AU-rich elements (AREs) mediate direct or indirect modulation of TNF-α, triggering mRNA degradation, translation inhibition, or translation activation of TNF-α mRNA [28–30]. Two recent studies have tried to explore the role of m^6^A in TNF-α production [31,32]. In human macrophages, the loss of m^6^A writer components eliminates m^6^A modification of TNF transcripts, thereby enhancing mRNA stability and TNF-α production [31]. Conversely, Tnf-α mRNA in murine macrophages was not the direct target of the m^6^A modification [32]. Unlike the commonly studied lipopolysaccharide (LPS)-induced inflammatory response, our study utilized T. gondii to infect THP-1 macrophages, and focused on the role of the m^6^A demethylase FTO. We found that after T. gondii infection, the upregulated expression of FTO attenuated the m^6^A modification of TNF transcripts, thereby enhancing the TNF mRNA stability and subsequently increasing TNF-α production. Specifically, we found that there were 9 potential m^6^A modification sites on human TNF-α mRNA. After T. gondii infection, the m^6^A modification levels at sites in the 5’UTR and 3’UTR regions of TNF-α mRNA decreased. Meanwhile, T. gondii infection significantly up-regulated the expression of the demethylase FTO in human macrophages. In FTO-knockdown cell lines, the m^6^A modification levels at sites in the 5’UTR region, especially in the 3’UTR region of TNF-α mRNA, did not significantly decrease after T. gondii infection. This suggests that the T. gondii infection-induced decrease in the m^6^A modification levels on TNF-α mRNA is associated with FTO. After T. gondii infection, the binding of TNF-α mRNA to the YTH domain family proteins decreased, suggesting that the degradation of TNF-α mRNA was reduced, which would result in increased TNF-α production. Our study elucidated, for the first time, the molecular mechanism of TNF-α expression in human macrophages induced by T. gondii infection from an m^6^A perspective.
In host-pathogen interactions, m^6^A modification exhibits a rapid onset of action and plays a crucial role in regulating downstream pathways [33,34]. On one hand, m^6^A modification affects pathogen gene expression by regulating the stability and translational efficiency of pathogen-derived mRNAs, thereby helping pathogens to escape host immune recognition [25,35,36]. On the other hand, m^6^A modification influences the expression of immune-related genes, and regulates the activation and differentiation of immune cells, thereby shaping host immune responses [22–24,32,37,38]. For example, after HIV-1 infection, the global m^6^A modification level of T cell transcriptome was significantly increased, and the immune response was subsequently changed [22]. In the case of T. gondii, while two recent studies reported the m^6^A methylation profiles of T. gondii tachyzoites and bradyzoites [19,39], our study, to the best of our knowledge, is the first to present the m^6^A landscape across the transcriptome of human macrophages after T. gondii infection.
Currently, research on m^6^A modification in the field of parasitology is limited to characterizing the m^6^A modification sites and modification levels of the parasites’ own RNA, identifying homologous m^6^A regulatory proteins, and focusing on the regulatory role of m^6^A modification in the parasite life cycle. For example, there are abundant m^6^A modifications on RNA of the bloodstream form and procyclic form of Trypanosoma brucei [40]. Subsequent studies reported that the m^6^A localized in the poly(A)-tailed mRNA of the variant surface glycoprotein (VSG) of Trypanosoma brucei inhibits RNA degradation by blocking deadenylation, thus ensuring the stability of VSG transcript [41,42]. PfMT-A70, pfYTH1 and pfYTH2 are m^6^A writer or reader proteins encoded by Plasmodium falciparum [43]. m^6^A marks are enriched near the 3’-boundary of T. gondii transcripts [39]. METTL3 and WTAP are essential in T. gondii viability and depletion of these two writer components prevents parasite replication [39]. Regarding m^6^A methylation differences among different T. gondii genotypes, pairwise comparisons of the RH strain (Type I), ME49 strain (Type II), and VEG strain (Type III) identified 735, 192, and 615 differentially methylated peaks (DMPs), as well as 172, 41, and 153 differentially methylated genes (DMGs) [19]. Enrichment analyses associated these DMPs and DMGs with cellular components (e.g., Golgi apparatus, plasma membrane) and signaling pathways (e.g., endocytosis, mTOR signaling), suggesting a role for m^6^A in the pathobiology of T. gondii.
It is well-known that T. gondii infection universally induces Th1 responses during the acute stage of the disease [44,45]. However, different strains of T. gondii induce varying types and levels of cytokines and signaling molecules, ultimately leading to distinct infection outcomes [44–46]. For example, after RH strain (type I) infection, macrophages exhibit only low levels of iNOS expression, resulting in negligible NO production; in contrast, following ME49 strain (type II) infection, macrophages express higher levels of iNOS and produce more NO [44]. Moreover, infection with different strains of T. gondii can induce macrophages to polarize into different subtypes [47]. It has been reported that Toxoplasma polymorphic effectors, such as ROP16 and GRA15, are involved in the regulation of these processes [47,48]. In this study, we investigated the regulatory role of m^6^A in the immune response of macrophages after infection with T. gondii RH strain. As different T. gondii strains trigger distinct immune responses in macrophages, besides Toxoplasma polymorphic effectors, whether m^6^A plays a role in the distinct host immune responses induced by different T. gondii strains is interesting and deserves further investigation.
In this study, we used T. gondii tachyzoites and human macrophages to investigate the role of m^6^A in innate immunity against infection. This model reflects the immune response of human macrophages in the acute stage of infection. From an m^6^A perspective, our findings reveal the impact of T. gondii, and potentially Apicomplexan protozoa, on public health. Notably, our findings are constrained to acute toxoplasmosis. In immunocompetent individuals, T. gondii infection is usually asymptomatic, existing in the form of bradyzoites. Therefore, the role of m^6^A in patients in the chronic stage or those with latent infection needs to be further investigated. Additionally, T. gondii infection in healthy populations has been linked to an increased risk of certain diseases, such as neuropsychiatric disorders, metabolic diseases, and autoimmune disorders [49–51]. An increasing number of studies have shown associations between m^6^A and these diseases [52–54]. However, whether a causal relationship exists among T. gondii infection, m^6^A alteration, and these diseases awaits further study.
In sum, this study provides new insights into the role of m^6^A modification in macrophage immune responses against T. gondii. Additionally, our findings uncover a new molecular mechanism for the immediate gene activation of TNF-α, through which m^6^A modification regulates TNF-α expression.
Supporting information
S1 TablePrimers used in this study.(DOCX)
S2 TablePrediction of m^6^A modification sites on human TNF-α mRNA.(DOCX)
S1 FigThe efficacy of FTO knock-down.A: qRT-PCR analysis of FTO knock-down efficacy. The mRNA abundance of FTO was measured by qRT-PCR. Data are shown as mean ± SD (*n *= 3). Statistical significance was determined using Student’s t-test. ***, P < 0.001. B: Western blotting analysis of FTO knock-down efficacy.(TIF)
S1 TextOriginal images for Western blotting.(DOCX)
S1 DataOriginal values used to build graphs.(XLSX)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lewis JM, Clifford S, Nsutebu E. Toxoplasmosis in immunosuppressed patients. Rheumatology (Oxford). 2015;54(11):1939–40. doi: 10.1093/rheumatology/kev 115 25969518 · doi ↗ · pubmed ↗
- 2Teimouri A, Mohtasebi S, Kazemirad E, Keshavarz H. Role of Toxoplasma gondii Ig G Avidity Testing in Discriminating between Acute and Chronic Toxoplasmosis in Pregnancy. J Clin Microbiol. 2020;58(9):e 00505-20. doi: 10.1128/JCM.00505-20 32321784 PMC 7448626 · doi ↗ · pubmed ↗
- 3Wang Q, Cao Y, Ye S, Ding M, Ge W, Liang Y, et al. Trem 2/Syk/PI 3K axis contributes to the host protection against Toxoplasma gondii-induced adverse pregnancy outcomes via modulating decidual macrophages. P Lo S Pathog. 2024;20(9):e 1012543. doi: 10.1371/journal.ppat.1012543 39250507 PMC 11412541 · doi ↗ · pubmed ↗
- 4Tian T, Wang M, Ma D. TNF-α, a good or bad factor in hematological diseases? Stem Cell Investig. 2014;1:12. doi: 10.3978/j.issn.2306-9759.2014.04.02 27358858 PMC 4923506 · doi ↗ · pubmed ↗
- 5El-Sayed NM, Ismail KA, Badawy AF, Elhasanein KF. In vivo effect of anti-TNF agent (etanercept) in reactivation of latent toxoplasmosis. J Parasit Dis. 2016;40(4):1459–65. doi: 10.1007/s 12639-015-0712-y 27876967 PMC 5118338 · doi ↗ · pubmed ↗
- 6Ricard J, Pelloux H, Pathak S, Pipy B, Ambroise-Thomas P. TNF-alpha enhances Toxoplasma gondii cyst formation in human fibroblasts through the sphingomyelinase pathway. Cell Signal. 1996;8(6):439–42. doi: 10.1016/s 0898-6568(96)00079-4 8958446 · doi ↗ · pubmed ↗
- 7Zhang Y, Chen H, Chen Y, Wang L, Cai Y, Li M, et al. Activated microglia contribute to neuronal apoptosis in Toxoplasmic encephalitis. Parasit Vectors. 2014;7:372. doi: 10.1186/1756-3305-7-372 25128410 PMC 4143554 · doi ↗ · pubmed ↗
- 8Qiu J, Xie Y, Shao C, Shao T, Qin M, Zhang R, et al. Toxoplasma gondii microneme protein MIC 3 induces macrophage TNF-α production and Ly 6C expression via TLR 11/My D 88 pathway. P Lo S Negl Trop Dis. 2023;17(2):e 0011105. doi: 10.1371/journal.pntd.0011105 36730424 PMC 9928027 · doi ↗ · pubmed ↗