Genomic and transcriptomic insights into the virulence and adaptation of shock syndrome-causing Streptococcus anginosus
Yu-Juan Lin, Chih-Ho Chen, Ian Yi-Feng Chang, Ruei-Lin Chiang, Hsing-Yi Wang, Cheng-Hsun Chiu, Yi-Ywan M. Chen

TL;DR
This study explores how Streptococcus anginosus adapts and causes infections by analyzing its genome and gene activity in different environments.
Contribution
The study reveals novel genomic and transcriptomic features of S. anginosus that explain its virulence and environmental adaptation.
Findings
The genome of strain KH1 contains nine genomic islands and phage-related proteins, suggesting horizontal gene transfer influences its evolution.
KH1 upregulates specific genes in saliva and serum, showing adaptation strategies for the oral cavity and bloodstream.
Despite lacking toxins, S. anginosus uses mechanisms like quorum quenching and glucan biosynthesis to survive and cause infections.
Abstract
Streptococcus anginosus is a common isolate of the oral cavity and an opportunistic pathogen for systemic infections. Although the pyogenic infections caused by S. anginosus are similar to those caused by Streptococcus pyogenes, S. anginosus lacks most of the well-characterized virulence factors of S. pyogenes. To investigate the pathogenicity of S. anginosus, we analysed the genome of a newly identified S. anginosus strain, KH1, which was associated with toxic shock-like syndrome in an immunocompetent adolescent. The genome of KH1 contains nine genomic islands, two Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated systems and many phage-related proteins, indicating that the genome is influenced by prophages and horizontal gene transfer. Comparative genome analysis of 355 S. anginosus strains revealed a significant difference between the sizes of the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
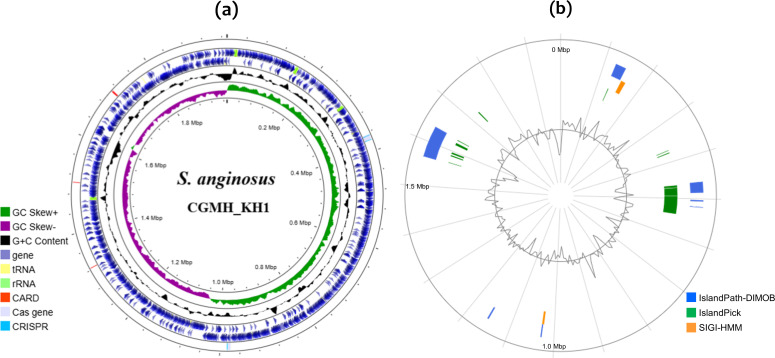 Fig. 1
Fig. 1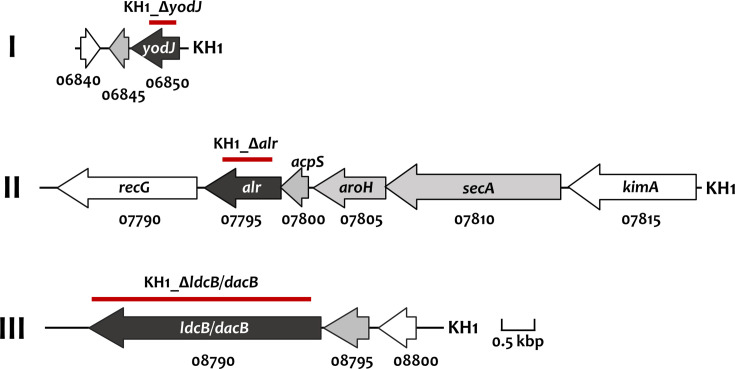 Fig. 2
Fig. 2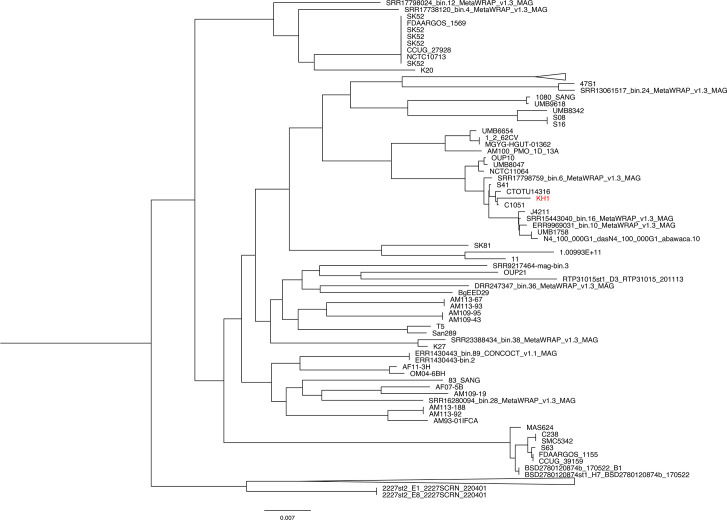 Fig. 3
Fig. 3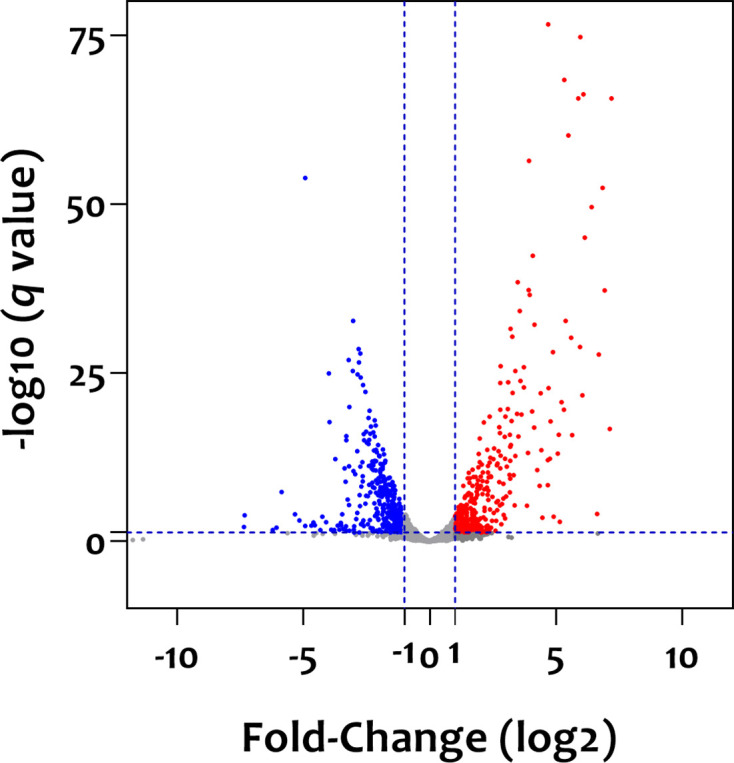 Fig. 4
Fig. 4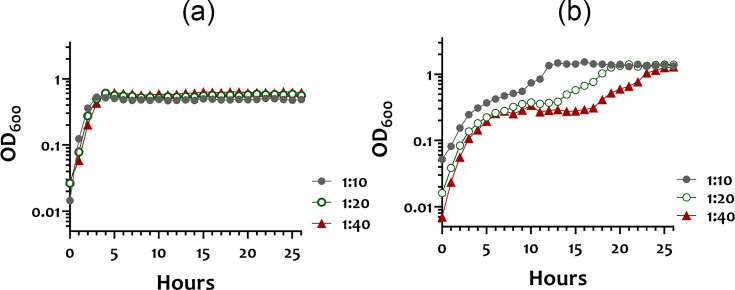 Fig. 5
Fig. 5| Locus tag* | Gene† | ARO‡ | Description | TPM | |
|---|---|---|---|---|---|
| SM§ | SV§ | ||||
|
| |||||
| PO908_06845 | – | – | AraC family ligand binding domain-containing protein | 15.0 | 15.8 |
| PO908_06850 |
| Putative carboxypeptidase YodJ | 41.3 | 33.6 | |
|
| |||||
| PO908_07795 |
| Alanine racemase | 506.1 | 152.1 | |
| PO908_07800 |
| – | Holo-[acyl-carrier-protein] synthase | 345.1 | 113 |
| PO908_07805 |
| – | Phospho-2-dehydro-3-deoxyheptonate aldolase | 349.4 | 156.7 |
| PO908_07810 |
| – | Translocase subunit SecA | 674.4 | 444.5 |
|
| |||||
| PO908_08790 |
| 307.8 | 102.6 | ||
| PO908_08795 | – | – | DUF6287 domain-containing protein | 91 | 49.2 |
| Island | Position ( | Locus tag | Total genes | %G+C content | Proposed functions | Identified by† |
|---|---|---|---|---|---|---|
| 1 | 129 037–133 832 | PO908_00725–00775 | 11 | 38.14 | Remnants of prophages | 1, 2 |
| 2‡ | 477 301–484 453 | PO908_02570–02635 | 14 | 34.41 | Remnants of prophages | 1, 2 |
| 3 | 501 193–513 503 | PO908_02750–02860 | 23 | 36.53 | Remnants of prophages | 1, 2 |
| 4‡ | 516 247–523 399 | PO908_02895–02960 | 14 | 34.41 | Remnants of prophages | 1, 2 |
| 5 | 1 025 622–1 030 166 | PO908_05375–05400 | 6 | 32.83 | Unknown | 1, 3 |
| 6 | 1 565 359–1 570 898 | PO908_08050–08085 | 8 | 38.23 | Toxin–antitoxin system | 1, 2 |
| 7 | 1 584 282–1 598 180 | PO908_08170–08220 | 11 | 33.22 | Conjugal transfer | 1, 2 |
| 8 | 1 612 764–1 615 030 | PO908_08300–08310 | 3 | 30.83 | Unknown | 1, 3 |
| 9§ | 1 615 732–1 635 653 | PO908_08320–08445 | 26 | 37.92 | Prophage-related proteins, toxin–antitoxin system and conjugal transfer | 1, 2 |
| Locus tag | Protein ( | Similarity* | Accession no.† | TPM | Fold-change | |
|---|---|---|---|---|---|---|
| SM‡ | SV‡ | SM/SV§ | ||||
| PO908_02850 | 129 | Single-stranded DNA-binding protein of | 48.5 | 15.6 | 3.1 | |
| PO908_02855 | 74 | Hypothetical protein of | 41.6 | 8.5 | 4.9 | |
| PO908_02860 | 41 | 33.8 | 7.8 | 4.3 | ||
| PO908_03035 | 69 | 0 | 0.3 | 0.0 | ||
| PO908_03040 | 226 | 63.4 | 76.1 | 0.8 | ||
| PO908_028225 | 532 | Abi family protein of | 25.3 | 24.9 | 1.0 | |
| PO908_028230 | 635 | ATP-binding protein of | 37 | 30.9 | 1.2 | |
| PO908_028235 | 392 | Hypothetical protein HMPREF9393_1502 of | 31.7 | 21.8 | 1.5 | |
| Locus tag | Gene* | Description | TPM | Fold-change | |
|---|---|---|---|---|---|
| SM† | SV† | SM/SV‡ | |||
|
| |||||
| PO908_00245 |
| Phosphoribosylformylglycinamidine cyclo-ligase | 204.9 | 19.6 | 10 |
| PO908_00250 |
| Phosphoribosylglycinamide formyltransferase | 195.1 | 19.5 | 10 |
| PO908_00315 |
| PTS-dependent dihydroxyacetone kinase | 457.2 | 30.2 | 15 |
| PO908_01385 |
| Ribose import-binding protein | 441 | 24.8 | 18 |
| PO908_01400 | – | Sugar-binding protein | 321.9 | 28.7 | 11 |
| PO908_03625 | – | Dipeptidase | 617.6 | 40.4 | 15 |
| PO908_03940 |
| 1,4- | 1384.5 | 34.7 | 40 |
| PO908_03945 |
| Glucose-1-phosphate adenylyltransferase | 2248.9 | 36 | 62 |
| PO908_03950 |
| Glycogen biosynthesis protein GlgD | 2043.8 | 34.5 | 59 |
| PO908_03955 |
| Glycogen synthase | 1360.4 | 23.7 | 57 |
| PO908_03960 |
| Glycogen phosphorylase | 951.9 | 21.3 | 45 |
| PO908_06745 |
| Glycerol dehydrogenase | 1390.1 | 11.4 | 122 |
| PO908_06750 |
| Fructose-6-phosphate aldolase | 1129 | 10.9 | 104 |
| PO908_06755 | – | Formate C-acetyltransferase/glycerol dehydratase family glycyl radical enzyme | 2152.9 | 14.7 | 146 |
| PO908_06760 |
| Lichenan permease IIC | 235.8 | 7.9 | 30 |
| PO908_06765 |
| PTS cellobiose-specific IIB | 440.2 | 8.9 | 49 |
| PO908_06770 |
| Lichenan-specific PTS IIA | 383.8 | 11.5 | 33 |
| PO908_07955 |
| Pyridoxine kinase | 1684.2 | 160.9 | 10 |
| PO908_08475 |
| Oligosaccharides import ATP-binding protein MsmX | 431.3 | 44.2 | 10 |
| PO908_09725 |
| Maltose maltodextrin-binding protein | 724.1 | 75.1 | 10 |
| PO908_10070 |
| Triosephosphate_isomerase | 234.2 | 2 | 117 |
|
| |||||
| PO908_01250 |
| ComG operon protein | 1367.2 | 30.3 | 45 |
| PO908_01255 |
| Competence protein CglB | 457.1 | 17.5 | 26 |
| PO908_01260 |
| Prepilin-type cleavage/methylation | 423.2 | 15.4 | 27 |
| PO908_01265 |
| Type II secretion system protein | 768 | 18.4 | 42 |
| PO908_01270 |
| Type II secretory pathway, pseudopilin PulG | 583.2 | 15.6 | 37 |
| PO908_01275 |
| Prepilin-type cleavage/methylation | 1435.9 | 20.4 | 70 |
| PO908_01280 |
| Competence protein | 864.6 | 17.8 | 49 |
| PO908_01380 |
| Single-stranded DNA-binding protein | 3270.2 | 24.3 | 135 |
| PO908_01395 |
| Transcriptional regulatory protein LnrK | 331.8 | 27.9 | 12 |
| PO908_02290 |
| ComF operon protein | 262.1 | 7.4 | 35 |
| PO908_02295 |
| Putative competence protein FC | 332.8 | 26.8 | 12 |
| PO908_03730 | – | Band-7–1 domain-containing protein | 175.8 | 11.8 | 15 |
| PO908_03735 | – | DNA-directed RNA polymerase subunit P | 319.8 | 15.2 | 21 |
| PO908_05005 |
| ComEA family DNA-binding protein | 756.6 | 12.2 | 62 |
| PO908_05010 |
| ComE operon protein | 345.2 | 10 | 35 |
| PO908_05500 |
| SAM-DrpA domain-containing protein | 620 | 9.4 | 66 |
|
| |||||
| PO908_00330 |
| Aquaporin | 335.8 | 25.3 | 13 |
| PO908_03520 |
| Lactococcin-A transport/processing ATP-binding protein LcnC | 166.4 | 14.6 | 11 |
| PO908_03560 |
| Putative Blp family class II bacteriocin | 430.8 | 10.9 | 40 |
| PO908_03570 |
| Putative bacteriocin Blp3.4 | 255.4 | 10 | 26 |
| PO908_03575 |
| Bacteriocin immunity protein | 121.9 | 8.4 | 15 |
| PO908_03580 |
| Hypothetical protein | 299.7 | 11.1 | 27 |
| PO908_03585 |
| Putative Blp family class II bacteriocin | 255.8 | 10 | 26 |
| PO908_10065 |
| UDP-glucose 4-epimerase | 624.3 | 4 | 156 |
|
| |||||
| PO908_00320 |
| PTS-dependent dihydroxyacetone kinase, ADP-binding subunit DhaL | 34.5 | 581.5 | 17 |
| PO908_00325 |
| PTS-dependent dihydroxyacetone kinase, phosphotransferase subunit DhaM | 26.1 | 344.6 | 13 |
| PO908_01790 | – | Hypothetical protein | 1155.5 | 17 | 68 |
| PO908_03515 | – | LAL-C2 domain-containing protein | 237 | 13.5 | 18 |
| PO908_03530 | – | Hypothetical protein | 244.5 | 11.4 | 21 |
| PO908_03590 | – | Putative bacteriocin transport accessory protein | 182.2 | 8.3 | 22 |
| PO908_03595 | – | Hypothetical protein | 486 | 16.6 | 29 |
| PO908_03740 | – | TPM domain-containing protein | 225.3 | 13.6 | 17 |
| PO908_05015 | – | Hypothetical protein | 185.9 | 9.2 | 20 |
| PO908_06190 | – | UPF0758 protein SpyM3_0777 | 191.6 | 10.1 | 19 |
| PO908_07950 |
| Integral membrane protein | 1418.1 | 91.6 | 15 |
| PO908_08610 | – | Hypothetical protein | 432.3 | 35.8 | 12 |
| PO908_08620 | – | HAD superfamily hydrolase | 213.9 | 19.5 | 11 |
| PO908_09650 |
| Putative competence-damage inducible protein | 1815.5 | 70.5 | 26 |
| PO908_10075 |
| Putative flavin reductase | 32.6 | 0.2 | 163 |
| PO908_10110 | – | Hypothetical protein | 2202.1 | 25.9 | 85 |
| Locus tag | Gene* | Description | TPM | Fold-change | |
|---|---|---|---|---|---|
| SM† | SV† | SM/SV‡ | |||
|
| |||||
| PO908_05530 |
| Acyl-CoA dehydrogenase | 18.7 | 193.4 | 0.10 |
| PO908_05555 |
| Acetyl CoA acetyltransferase | 8.9 | 87.6 | 0.10 |
| PO908_05560 | – | 3-Hydroxybutyryl-CoA dehydrogenase | 9.1 | 145 | 0.06 |
| PO908_07025 |
| Galactose 6 phosphate isomerase subunit | 0.9 | 11.9 | 0.08 |
| PO908_07040 |
| DEAD box ATP-dependent RNA helicase CshA | 18.4 | 561.5 | 0.03 |
| PO908_08785§ |
| Pyruvate oxidase | 41.3 | 341.5 | 0.12 |
|
| |||||
| PO908_00045 | – | Putative cell division protein DivIC | 8.9 | 84.5 | 0.11 |
| PO908_07195 | – | FtsX-like permease family protein | 2 | 27.3 | 0.07 |
|
| |||||
| PO908_00040 | – | RNA-binding S4 domain-containing protein | 1 | 17.3 | 0.06 |
| PO908_00775 |
| Tyrosine recombinase XerC | NE | 17.4 |
|
| PO908_05535 | – | LysR family transcriptional regulator | 4.8 | 75.7 | 0.06 |
| PO908_06815 |
| Threonine tRNA ligase | 63.9 | 631.6 | 0.10 |
|
| |||||
| PO908_00020 |
| Putative lipoprotein YehR | 4.6 | 50.5 | 0.09 |
| PO908_00780 | – | Hypothetical protein | NE | 22.8 |
|
| PO908_00840 | – | Hypothetical protein | NE | 19.2 |
|
| PO908_05525 |
| Putative quorum-quenching lactonase YtnP | 8.2 | 82.8 | 0.10 |
| PO908_06020 | – | Hypothetical protein | 2 | 22.8 | 0.09 |
| PO908_09935 |
| Chromosome partition protein Smc | 7.9 | 465.5 | 0.02 |
- —http://dx.doi.org/10.13039/501100005795 Chang Gung Memorial Hospital, Linkou
- —http://dx.doi.org/10.13039/100020595 National Science and Technology Council
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsStreptococcal Infections and Treatments · Infective Endocarditis Diagnosis and Management · Oral microbiology and periodontitis research
Data Availability
The data supporting the findings are available within the article or supplementary files. The raw RNA-seq reads have been deposited in the NCBI GEO database under the assigned accession number GSE285825. The accession number of the Streptococcus anginosus KH1 genome is CP117042.1. The accession number of the raw reads of KH1 transcriptomes is GSE285825.
Introduction
Streptococcus anginosus, a member of the S. anginosus group (SAG) [1], is commonly found in the mucosal membranes of the oral cavity [2]. All species of SAG, i.e. S. anginosus, Streptococcus constellatus and Streptococcus intermedius, can spread from the oral cavity to the upper respiratory, digestive and reproductive tracts and are considered part of the normal microbiome. However, SAG species have been frequently implicated in pyogenic infections [3], which are characterized by acute inflammatory responses and pus formation. For instance, a population-based surveillance study in Canada revealed that the incidence of SAG infections was higher than those caused by Streptococcus pyogenes and Streptococcus agalactiae [4]. More recent research further confirmed that bacteremia caused by SAG could result in significant morbidity and mortality [5]. The suppurative infections caused by SAG resemble those caused by S. pyogenes, including pharyngitis, bacteremia and serious deep tissue infections [4,68], indicating that the pathogenic potential of SAG may be greater than previously expected. Although not all SAG infections result in pyogenic outcomes [9], recent case reports and reviews also suggest that SAG species are common pathogens in conditions such as descending necrotizing mediastinitis [10], intracranial infections [1112] and even osteomyelitis [13]. Furthermore, studies have shown that S. anginosus could co-exist with cystic fibrosis (CF)-associated Pseudomonas aeruginosa DWW2 in biofilms and enhance P. aeruginosa DWW2 pyocyanin production*,* resulting in increased virulence in the infection mode [1415]. Although the role of S. anginosus in CF pathogenesis requires further investigation, this study suggests that S. anginosus could modulate the virulence of cohabitating microbes. Lastly, a recent study identified an enrichment of S. anginosus in the gastric microbiome of patients with gastric cancer, and the enrichment is inversely correlated with the presence of Helicobacter pylori [16]. The association between S. anginosus and gastric tumorigenesis was further investigated in a mouse model, confirming the pathogenic potential of S. anginosus [17].
While the clinical significance of SAG is evident, the virulence mechanisms of SAG remain poorly understood [2]. To date, the few studied virulence factors of S. anginosus include a laminin-binding protein that mediates the adhesion of S. anginosus to the exposed basement membrane of heart valve tissues during the course of endocarditis [18], a fibronectin-binding protein that enhances the interaction of S. anginosus F4 with polymorphonuclear leukocytes [19], and homologues of S. pyogenes streptolysin S, a haemolysin produced through the sag operon expression [2021]. However, not all S. anginosus strains possess the sag cluster. To be noted, genes encoding the streptococcal pyrogenic exotoxins, which are implicated in invasive infections, are absent in * S. anginosus*. Though, studies have shown that the LuxS-based, autoinducer-II signalling pathway regulates biofilm formation in S. anginosus [22] and that S. anginosus exhibits antibiotic resistance [23], suggesting that, similar to other streptococcal species, virulence expression is regulated by environmental growth conditions in S. anginosus.
Genomic studies have revealed that the CRISPR-Cas systems, prophages and mobile genetic elements are present among * S. anginosus* strains [2426]. Furthermore, S. anginosus exhibits natural competence and, thus, can acquire virulence genes, such as antibiotic resistance genes, through horizontal gene transfer (HGT). For instance, it has been hypothesized that S. anginosus strain 47S1 acquires genes encoding resistance to tetracycline, aminoglycosides, fusidic acid and fluoroquinolone via HGT [24]. The presence of vanG, encoding vancomycin resistance, has also been described in S. anginosus SA1 [27]. In addition, a comparative genomic study of S. anginosus JA4206 revealed unique genes and variants that could contribute to the acquisition of daptomycin resistance [26].
A strain of S. anginosus, designated KH1, was recently isolated from a previously healthy, young patient at Kaohsiung Chang Gung Memorial Hospital [28]. This patient presented with streptococcal toxic shock-like syndrome 2 days following a dental procedure for caries, suggesting that KH1 likely originated in the oral cavity. Although a narrow haemolytic zone developed around the colony on a blood agar plate [28], the sag genes were not identified by PCR. To investigate the activity that enables KH1 to establish systemic infections, we determined the genome and analysed the transcriptomes of KH1 cultivated in environments mimicking the oral cavity and the bloodstream. The genomic analysis identified nine genomic islands, reflecting its natural competence. We also found that the transcriptomes were significantly regulated by growth media, indicating that S. anginosus could modify its transcriptome to facilitate survival in specific niches.
Methods
Bacterial strains and growth conditions
S. anginosus KH1 was routinely cultivated in Todd Hewitt (TH, Difco) broth, at 37 °C in a 10% CO_2_ atmosphere. For transcriptomic analysis, KH1 was cultivated in either artificial saliva medium (SV) or modified serum medium (SM). The artificial SV was prepared as per Wong and Sissons [29], with some modifications, and contains 0.25% mucin, 1% proteose peptone, 1% trypticase peptone, 1% yeast extract, 0.25% KCl and 0.25% glucose. To prepare the modified SM, venous blood samples were collected from healthy human volunteers in BD Vacutainer™ SST™ II Advance Tubes (BD). The blood samples were subjected to centrifugation at 2500 g, 25 °C for 10 min to remove the blood cells. The recovered plasma was heated at 56 °C for 30 min to inactivate complement. HEPES and glucose were added to the plasma to final concentrations of 40 mM and 0.5%, respectively, to make modified SM.
To determine growth kinetics, overnight cultures of KH1 were grown in TH broth and subcultured into pre-warmed artificial SV or modified SM at 1 : 10, 1 : 20 and 1 : 40 dilutions. The growth at OD_600_ was monitored using a Bioscreen C Microbiology Reader (Oy Growth Curve AB Ltd.). For each condition, three biological replicates were analysed.
Purification of chromosomal DNA, sequencing and annotation
Total cellular DNA was isolated from a mid-exponential (OD_600_=0.6) phase culture of S. anginosus KH1 as described previously [30]. Genome sequencing was performed by a commercial service (Genomics, Taiwan). Briefly, the genomic DNA was sheared using g-TUBE (Covaris), and the library was prepared using the SMRTbell express template Prep Kit 2.0 (PacBio). The PacBio Sequel/Sequel II platform was used to determine the genome sequence. A primary filter analysis was performed on the Sequel instrument to trim the low-quality regions, and the secondary analysis was performed using the SMRT analysis pipeline version 8.0 (https://www.pacb.com/support/software-downloads/). The reads were assembled initially using Hifiasm v0.8 with the default setting [31]. The contigs were further assembled using SSPACE-Long v1.1 [32]. ORFs were predicted and annotated using Prokka v1.12 (https://github.com/tseemann/prokka). Clusters of Orthologous Groups (COGs) were analysed using eggNog-mapper 2.1.12 [33], and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway was used to predict gene functions. Genomic islands and CRISPR systems were identified through IslandViewer 4 [34] and CRISPRCasFinder 4.2.20 [35], respectively. Putative antibiotic resistance loci were analysed by Comprehensive Antibiotic Resistance Database (CARD) [36]. The complete genome sequence of S. anginosus KH1 has been deposited in the GenBank database with the accession number CP117042.1.
RNA isolation and transcriptomic analysis
Total cellular RNA was isolated from KH1 cultures at OD_600_=0.3 as described [37] and further purified using the RNeasy kit (Qiagen). rRNA was removed from the RNA preparation using RiboMinus transcriptome isolation kits (Invitrogen). PCR was performed with primers specific for PO908_05840 to ensure that the preparations were free of chromosomal DNA. Transcriptome sequencing was performed by a commercial service (Welgene, Taiwan). Briefly, the RNA library was constructed using a SureSelect XT HS2 mRNA library preparation kit (Agilent, USA). The sequences were determined using Illumina’s sequencing-by-synthesis technology (Illumina, USA). Reads were assessed through a Phred quality score (Q score of 20) and trimmed using Trimmomatic v0.36 [38]. The trimmed reads were mapped to the genome sequence of KH1 by using HISAT2 [39]. TPM (transcripts per million) was used to calculate the expression level of each gene. The significant difference in the expression level of each locus between the two growth conditions was analysed using DESeq2 v1.28.1 [40] with genome bias detection/correction and Welgene Biotech’s in-house pipeline. For each condition, duplicate samples were analysed. Genes exhibiting a fold change >2.0 and a q-value <0.05 between the two culture conditions were considered significant. The raw RNA-seq reads have been deposited in the NCBI GEO database under the accession number GSE285825.
Comparative genomic analysis
We used panaroo (v1.5.0) [41] to generate the pan-genome of 355 S. anginosus strains (Table S1, available in the online Supplementary Material). Among these genomes, gene annotation files of 275 genomes were downloaded from the NCBI database (accessed on 19 July 2024). For 79 incomplete genomes without gene annotation files (accessed on 19 July 2024), we predicted and annotated the incomplete genome by Prokka [42]. The phylogenetic tree of these strains and KH1 was generated using iqtree2 (v2.1.2) [43]. The core genome contains orthologues from all strains with ≥99% identity.
Construction of recombinant S. anginosus strains
Recombinant S. anginosus strains deficient in yodI, alr and lacB were generated by ligation mutagenesis [44]. In short, two DNA fragments flanking the target region were amplified from S. anginosus KH1 by PCR using specific primers. The resulting products were restriction-digested and mixed with DNA fragments containing a nonpolar erythromycin (Em) resistance gene (erm) [45] in a ligation reaction. The ligation mixture was introduced into KH1 by natural transformation. The allelic exchange event in the Em-resistant transformants was verified by colony PCR with primers located outside the insertion site of the erm gene. All primers used in mutant construction are listed in Table S2.
Determination of MIC and minimal bactericidal concentration
The MIC for vancomycin in S. anginosus strains was determined using a 96-well plate assay, following Clinical and Laboratory Standards Institute guidelines. The plates were incubated at 37 °C under 10% CO_₂_ for 24 h. The minimal bactericidal concentration (MBC) was subsequently assessed by subculturing samples from wells without visible growth onto TH agar plates to determine the lowest bactericidal concentration. For each strain, three biological replicates were analysed.
Results and discussion
The general features of the S. anginosus KH1 genome
The general features of the KH1 genome are listed in Fig. 1. Briefly, KH1 harbours a circular chromosome of 1 970 657 bp, with a G+C content of 38.81 mol%. The complete genome contains 2026 predicted CDs, including 12 rRNA in four operons, 61 tRNA and 3 ncRNA. Two CRISPR-Cas systems were identified in the genome by CRISPRCasFinder [35]. Based on blast analysis, one belongs to type II-A of the class II CRISPR-Cas systems, and the other belongs to type III-A of the class I systems. While type II-A is commonly found in S. anginosus [46], both types II-A and III-A are dominant subtypes in streptococci [47]. In line with other S. anginosus genomes, putative phage-related genes were annotated in the genome. The presence of CRISPR-Cas systems and phage proteins suggests that the genome of KH1 is influenced by HGT.
Genome structure of S. anginosus KH1. (a) Circular representation of the chromosome. The genome position, scaled in Mbp, from base 1 is shown on the inner circle. From inner to outer circles, the second circle shows the GC skew curve (10 kbp window and 0.1 kbp incremental shift). The values for plus and minus strands are shown in green and purple, respectively. The third circle shows the G+C content (in 10 kb window, black). The values that are greater than, and below, the average (38.81%) are shown. The fourth circle shows the CDs. Genes are in light purple. tRNA and rRNA are in yellow and mint, respectively. The outermost circle shows loci predicted by CARD (red), the predicted cas genes (light lavender) and CRISPRs (sky blue). (b) Circular representation of the predicated genomic islands. The inner circle displays the G+C content. The middle circles indicate loci predicted by IslandPath-DIMOB (cobalt blue), IslandPick (green) and SIGI-HMM (orange).
Similar to other S. anginosus genomes, loci involved in metabolism (COG categories C, E, F, G, H, I, P and Q) and information processing and storage (COG categories J, K and L) are the most abundant categories, comprising 31.7 and 24.6% of the genome, respectively (Table S3). This genome also harbours more than 500 poorly characterized loci, covering 28.4% of the genome. A relatively high percentage (10.35%) of hypothetical proteins was observed in the KH1 genome, compared to a subset of 13 complete S. anginosus genomes (7.2–12.0%), which included strains that are phylogenetically distant from or relatively close to KH1 (see below for a detailed description of the phylogenetic analysis). The proportion of hypothetical proteins in S. anginosus KH1 is notably higher than that in S. pyogenes A20 (CP003901, 8.8%), isolated from a patient with necrotizing fasciitis [48]. Interestingly, the KH1 genome features a duplicated region of about 39 kbp. The duplicated genes exhibit point mutations, resulting in ORF variations. One region contains the PO908_02445–02760 CDs, while the other contains the PO908_02765–03075 CDs. To be noted, the 39 kbp duplicated region also contains a genomic island coding for 14 genes; thus, the KH1 genome contains two identical islands, designated island 2 (PO908_02570–02635) and island 4 (PO908_02895–02960). The genomic island in the duplicated region is described further below.
Putative antibiotic resistance loci
Three putative vancomycin resistance loci, yodJ, alr and ldcB/dacB, were predicted in the KH1 genome by CARD [36] (Table 1). Based on the distance between the flanking genes, it is predicted that both yodJ and ldcB/dacB are part of two-gene operons (operons I and III), and alr is the last gene of a four-gene operon (operon II, Fig. 2). The predicted two-gene operon containing yodJ and PO908_06845 (operon I) encodes a putative carboxypeptidase and an AraC family ligand binding domain-containing protein, respectively. PO908_06845 does not possess a helix-turn-helix DNA binding domain, and thus, its function in the expression regulation of operon I is unknown. Transcriptomic analysis of operon I demonstrated low expression levels for its constituent genes in either the artificial SV or the modified SM (Table 1).
Schematic representation of the putative antibiotic resistance loci in S. anginosus KH1. The relative location and orientation of each ORF are shown. The proposed gene name based on blastp analysis is indicated within or above the gene. The PO908_tag number is listed below the gene. Genes identified by CARD are in black, and loci predicated to be in the same operon are in grey. The regions replaced by non-polar erm in the mutant strains are indicated by a thick red line and listed above the gene cluster.
Operon II consisting of four genes, secA-aroH-acpS-alr, encodes translocase SecA, an aldolase, holo-[acyl-carrier-protein] synthase and an alanine racemase, respectively. Expression levels of the operon II genes were elevated by 1.5–3-fold when the organism was grown in modified SM, compared to expression derived from growth in artificial SV.
Lastly, operon III, PO908_08795-ldcB/dacB, encodes a hypothetical protein and a putative ld-carboxypeptidase, respectively. The putative LdcB/DacB contains a YSIRK-type signal peptide at the N-terminus, three bacterial SH3 domains and four group B Streptococcus protein-like domains in the central part, followed by a conserved carboxypeptidase domain of the LdcB superfamily. Similar to operon II, the expression of this operon was also upregulated when the organism was grown in the modified SM, compared to the artificial SV.
An attempt was made to investigate whether these genes are involved in vancomycin resistance using isogenic mutant strains. S. anginosus KH1 is very sensitive to vancomycin, with a MIC at 1 μg ml^−1^ and a MBC at 64 μg ml^−1^. Inactivation of yodJ, alr or ldcB/dacB did not increase the sensitivity of S. anginosus KH1 to vancomycin significantly. All mutant strains exhibited the same MIC and MBC as WT KH1. Thus, the functions of these genes in antibiotic resistance are unclear.
Genomic islands
The genomic islands were predicted using IslandViewer 4 (Fig. 1b), which incorporates IslandPath-DIMOB [49], IslandPick [34] and SIGI-HMM [50]. Regions identified by at least two algorithms were recognized as an island. This process resulted in the discovery of nine genomic islands (Tables 2 and S4–S11). All islands exhibited a G+C content lower than the overall genome (38.81%), thereby confirming their origin by HGT. Although many proteins encoded by genes on the islands are functionally unknown, most of the islands harbour loci encoding phage proteins, proteins related to conjugal transfer or a toxin–antitoxin system. The expression of most genes on islands 2, 3 and 4 (Tables S5 and S6) was upregulated in KH1 cultures grown in the modified SM; however, as most of the loci were of unknown function, the role of these proteins for growth in the bloodstream remains unknown. It was also observed that most of the genes on islands 1 and 5 (Tables S4 and S7), which include phage proteins (PO908_00730 and 00765 of island 1) and a putative TonB-dependent receptor (PO908_05375 of island 5), exhibited enhanced expression in the artificial SV, compared to expression in the modified SM, suggesting that these genes may play a role in adaptation to the oral cavity environment. The impact of culture medium on the expression of loci located on islands 6, 7, 8 and 9 was less straightforward (Tables S8–S11). To be noted, expression of loci encoding proteins related to the toxin–antitoxin system, i.e. PO908_08055 of island 6 and PO908_08330, 08365 and 08435 of island 9, did not appear to be altered by the bacterial growth medium, suggesting that these proteins may be required in both the oral cavity and in the bloodstream.
Pan genome construction and phylogenetic analysis
Upon analysing 355 accessible S. anginosus genomes, we found that the pan genome is comprised of 8316 genes, including 909 core genes (99%≤strains≤100%), 368 soft core genes (95%≤strains<99%), 1083 shell genes (15%≤strains<95%) and 5956 cloud genes (0%≤strains<15%). The large difference between the number of core and pan genomes indicates a significant variation among strains. Phylogenetic analysis unveiled three major clades, with one clade encompassing the majority of the strains, including KH1 (Figs 3 and S1). KH1 is phylogenetically closest to strain C1051 [51], which is also a blood isolate. According to a comparative genomic study by Prasad et al., KH1 belongs to the group 1 genotype [52], which encompasses isolates from various human body sites, with the exception of urinary isolates, which is in agreement with the isolation origin of KH1.
Phylogenetic analysis of S. anginosus KH1 and other S. anginosus strains. The tree was constructed from 355 S. anginosus genomes by iqtree2 (v2.1.2). Detailed information on the collapsed branches is listed in Fig. S1.
Through pan genome analysis, eight unique genes were identified in KH1 (Table 3). Although the involvement of these unique genes in KH1 pathogenicity cannot be predicted based on annotation, it was noted that three unique genes exhibited a greater than threefold increase in expression when cultivated in the modified SM, compared to the artificial SV. Thus, it is tempting to suggest that variations at the genetic and expression levels among S. anginosus strains could affect the ability of the strain to transit from the oral cavity to the bloodstream, as demonstrated in Escherichia coli [53], and that the expression of unique genes may play a role in the survival of KH1 in the bloodstream.
Genes encoding proteins involved in purine and glycogen biosynthesis, competence and putative bacteriocin production were induced during growth in modified SM
As elevated gene expression in the modified SM, compared to expression levels in the artificial SV, may suggest the necessity of a particular gene for survival in the bloodstream, we sought to identify essential virulence genes by comparing transcriptomes of S. anginosus KH1 derived from growth in either of these conditions. Indeed, different transcriptomes were expressed by S. anginosus grown in these two types of media, reflecting altered gene expression in differing environments (Fig. 4). To be noted, loci exhibiting a TPM of less than ten were not included in the analysis as the low expression levels may not contribute significantly to the overall activity of KH1 under a specific growth condition. Three key findings resulted from the analysis of loci whose expression was elevated by ≥10-fold in the serum-like medium (Table 4). First, in agreement with an observation in S. sanguinis SK36 [54], most loci of the pur operon (PO908_00230–00300), responsible for purine biosynthesis, were significantly upregulated in the modified SM. Although the precise role of purine biosynthesis in pathogenesis remains undefined, studies in Staphylococcus aureus and Enterococcus faecalis indicated that induction of the de novo purine biosynthesis pathway is critical for the adaptation of these two species to growth in biofilms [55]. Additionally, a purine nucleotide transporter protein (TMPC) has been shown to act as a ligand for the adherence and invasion of gastric epithelial cells by S. anginosus ATCC33397 via TMPC-ANXA2 interaction [17]. This study further demonstrated that TMPC-ANXA2 interaction can activate the Mitogen-Activated Protein Kinase signalling pathway of the infected cells, leading to gastric inflammation, atrophy and even tumorigenesis in mice. Although we did not observe altered expression of the TMPC homologue in KH1 (PO908_05780) in our culture conditions, upregulation of purine biosynthesis suggests that this pathway plays a role in the survival of S. anginosus in hosts.
Volcano plot demonstrating the impact of culture medium on S. anginosus KH1 transcriptomes. Red and blue dots represent loci with a fold-change of ≥2 and ≤ 0.5 in modified SM compared to artificial SV, respectively. Loci with fold-changes between 0.5 and 2 or without significant expression changes are shown in grey.
Second, though the modified SM is nitrogen-rich, it was surprising to find that genes involved in glycogen biosynthesis (PO908_03940–03960) were upregulated by ~40- to 60-fold. Nonetheless, a study [56] in Shigella flexneri found that intracellular S. flexneri expresses a higher level of glycogen branching enzyme (GlgB) and glucose-1-phosphate adenylyltransferase (GlgC), compared to its extracellular counterpart, suggesting that glycogen production under glucose-limiting conditions could optimize the fitness of intracellular S. flexneri. Thus, it is possible that the upregulation of glycogen production in the modified SM is not for carbon nutrient storage, but rather that S. anginosus employs glycogen biosynthesis to facilitate its transition from the oral cavity to the bloodstream via an unknown mechanism.
Third, genes associated with both competence development (PO908_01250–01280, 02290–02295 and 05005–05010) and bacteriocin production (PO908_03560–03595) were upregulated in the modified SM, suggesting that these two systems are sensitive to the same environmental cues. The coordinate regulation of competence and bacteriocin production has also been described in Streptococcus pneumoniae [57], in which the competence peptide could induce the expression of pneumocin. It is proposed that such regulation allows S. pneumoniae to gain additional genotypes, such as antibiotic resistance, from competing bacteria. Thus, it is possible that upregulation of bacteriocin production and acquisition of extracellular DNA could optimize the fitness of S. anginosus in the bloodstream.
Expression of loci involved in quorum quenching and translation was elevated in the artificial SV
A total of 377 loci exhibited higher expression when grown in the artificial SV than the modified SM, though the level of increase was less pronounced compared to upregulation in the modified SM. Specifically, 13 loci showed a ≥10-fold increase in expression in the artificial SV (Table 5), compared to 61 loci in the modified SM (Table 4). Three key observations were noted. First, the expression of a putative quorum-quenching synthesis system, including a LysR family regulator (PO908_05535), an acyl-CoA dehydrogenase (PO908_05530) and a putative lactonase YtnP (PO908_05525), was upregulated more than tenfold. Lactonase YtnP is able to interrupt bacterial signalling based on acyl homoserine lactones and has been identified in both Gram-positive and Gram-negative bacteria. Specifically, YtnP of Bacillus paralicheniformis ZP1 inhibits biofilm formation and breaks down mature P. aeruginosa PAO1 biofilms [58]. As the putative YtnP is a cytoplasmic protein, it is possible that the release of lactonase from lysed S. anginosus could optimize the competitiveness of the remaining * S. anginosus* population in the oral niche by interrupting the quorum sensing regulation of other bacteria in the same niche.
Second, genes encoding several enzymes involved in ribosome biosynthesis, translation and RNA decay (PO908_00040, 06815 and 07040) and cell division (PO908_00045 and 07195) showed higher expression in the artificial SV compared to the modified SM. In contrast, transcription of these groups of genes was not affected by growth in the modified SM, suggesting that S. anginosus adapted to the artificial SV more efficiently. Indeed, when a culture of S. anginosus KH1 grown in TH broth overnight was subcultured into artificial SV, we observed comparable growth kinetics to a culture grown in TH, with a generation time of 45 min and a final yield at OD_600_≈0.6, in all inoculum dilutions tested (Fig. 5a). In contrast, KH1 exhibited a diauxic growth pattern when subcultured into the modified SM, where the length of the transition period was more evident at a lower inoculum size (Fig. 5b). KH1 exhibited a generation time of 2 h and a final OD_600_ of 0.1 in the first growth phase, but the final yield achieved an OD_600_=1 in all dilutions tested, suggesting that KH1 is equipped to persist in the bloodstream.
The growth of S. anginosus KH1 in artificial SV (a) and modified SM (b). Overnight cultures of KH1 grown in TH medium were diluted into the respective medium at 1 : 10, 1 : 20 or 1 : 40. The presented result is the representative of three biological replicates.
Finally, the expression of pyruvate oxidase (PO908_08785) in artificial SV, an enzyme catalysing H_2_O_2_ production, was eightfold higher than that in the modified SM, suggesting that H_2_O_2_ production could provide a competitive advantage for S. anginosus in the oral cavity, as has been reported previously in S. sanguinis, which can antagonize Streptococcus mutans by inflicting oxidative damage via reactive oxygen species [59].
Conclusions
The genome of S. anginosus KH1 is shaped by chromosomal duplication and HGT events, and the large size of the pan genome reflects the variations among S. anginosus strains. The environmentally dependent differential expression of specific loci examined in this study strongly suggests that S. anginosus KH1 is equipped to maintain a niche in the oral cavity and to survive in the bloodstream. The genome characterization and transcriptomic analysis of S. anginosus KH1 described here provide insights into the physiology and pathogenic potential of this bacterium.
Supplementary material
10.1099/mic.0.001535Uncited Fig. S1.
10.1099/mic.0.001535Uncited Supplementary Material 1.
10.1099/mic.0.001535Uncited Supplementary Material 2.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jensen A Hoshino T Kilian M Taxonomy of the Anginosus group of the genus Streptococcus and description of Streptococcus anginosus subsp. whileyi subsp. nov. and Streptococcus constellatus subsp. viborgensis subsp. nov Int J Syst Evol Microbiol 2013632506251910.1099/ijs.0.043232-023223817 · doi ↗ · pubmed ↗
- 2Asam D Spellerberg B Molecular pathogenicity of Streptococcus anginosus Mol Oral Microbiol 20142914515510.1111/omi.1205624848553 · doi ↗ · pubmed ↗
- 3Pilarczyk-Zurek M Sitkiewicz I Koziel J The clinical view on Streptococcus anginosus group - opportunistic pathogens coming out of hiding Front Microbiol 20221395667710.3389/fmicb.2022.95667735898914 PMC 9309248 · doi ↗ · pubmed ↗
- 4Laupland KB Ross T Church DL Gregson DB Population-based surveillance of invasive pyogenic streptococcal infection in a large Canadian region Clin Microbiol Infect 20061222423010.1111/j.1469-0691.2005.01345.x 16451408 · doi ↗ · pubmed ↗
- 5Laupland KB Pasquill K Parfitt EC Dagasso G Steele L Streptococcus anginosus group bloodstream infections in the western interior of British Columbia, Canada Infect Dis 20185042342810.1080/23744235.2017.141616329262754 · doi ↗ · pubmed ↗
- 6Babbar A Kumar VN Bergmann R Barrantes I Pieper DH et al Members of a new subgroup of Streptococcus anginosus harbor virulence related genes previously observed in Streptococcus pyogenes Int J Med Microbiol 201730717418110.1016/j.ijmm.2017.02.00228274699 · doi ↗ · pubmed ↗
- 7Whiley RA Hall LM Hardie JM Beighton D A study of small-colony, beta-haemolytic, Lancefield group C streptococci within the anginosus group: description of Streptococcus constellatus subsp. pharyngis subsp. nov., associated with the human throat and pharyngitis Int J Syst Bacteriol 1999491443144910.1099/00207713-49-4-144310555325 · doi ↗ · pubmed ↗
- 8Junckerstorff RK Robinson JO Murray RJ Invasive Streptococcus anginosus group infection-does the species predict the outcome?Int J Infect Dis 201418384010.1016/j.ijid.2013.09.00324215937 · doi ↗ · pubmed ↗