Genomic analysis of Legionella pneumophila in the drinking water system of a large building over 25 years
Helena M. B. Seth-Smith, Adrian Egli, Sylvia Gautsch, Michael M. Bornstein, Eva M. Kulik

TL;DR
This study tracks Legionella pneumophila in a building's water system over 25 years, finding a single lineage and recommending genomic analysis for monitoring.
Contribution
The study provides a 25-year genomic analysis of L. pneumophila in a building's water system, revealing a single lineage and insights into its evolution.
Findings
L. pneumophila was detected in 38.8% of 309 water samples over 25 years.
All isolates were sequence type 45 and closely related with only 408 SNPs after recombination removal.
A single lineage of L. pneumophila colonized the building's water system, with phylogeny suggesting good circulation and recolonization after cleaning.
Abstract
Legionella pneumophila, the causative agent of Legionnaires’ disease, is often found in the plumbing systems of buildings, from where it can be transmitted to humans via inhalation or aspiration of contaminated water drops. Annual routine water sampling from the potable water system of an occupational healthcare building in Basel over 25 years was performed in accordance with national guidelines. Overall, 309 water samples were collected at 38 time points over the period of 25 years. L. pneumophila was recovered from 120 water samples (38.8%) from 26 time points. No clinical infections were recorded during this period. Initial decontamination measures were successful for ~12 years, after which an increase in the total number of Legionella c.f.u. as well as of L. pneumophila-positive sites was noticed in 2007. Whole genome sequencing (WGS) analysis of n=123 isolates from n=113 samples…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
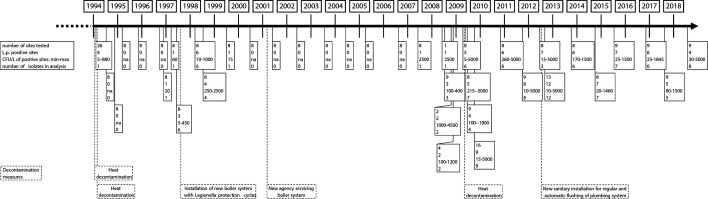 Fig. 1
Fig. 1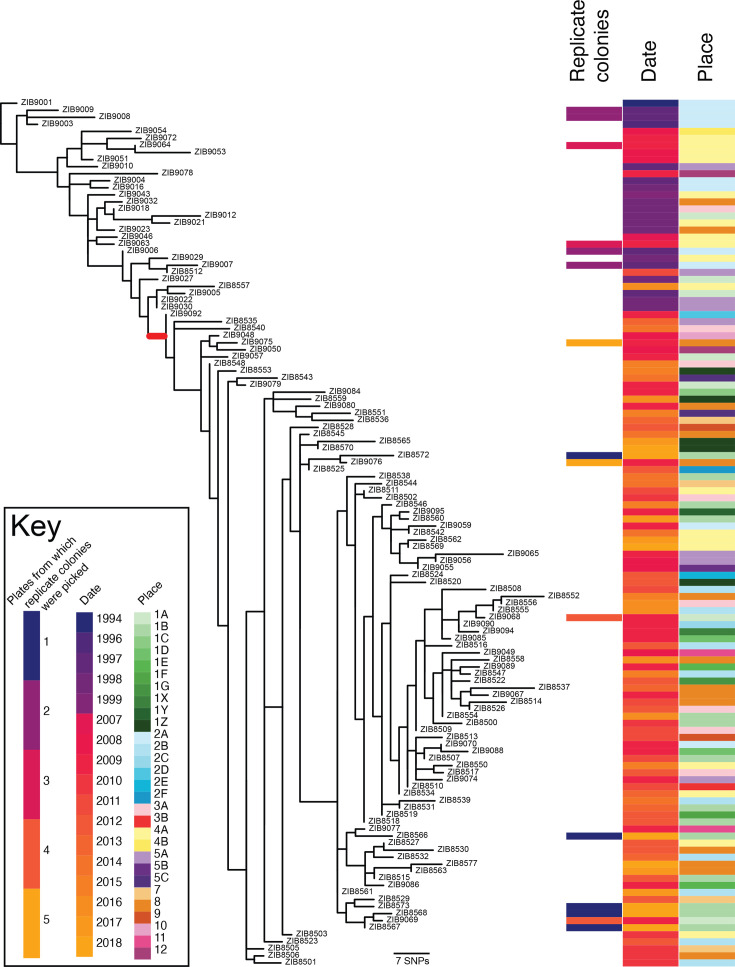 Fig. 2
Fig. 2| Start | End | Length | Found in | Match to strain accession | Comment | ||
|---|---|---|---|---|---|---|---|
| 57,502 | 60,351 | 2,850 | All ST45 genomes | 100.00 | 99.79 | Lorraine | Homology to ISLpn9 |
| 65,009 | 66,795 | 1,787 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 69,573 | 75,226 | 5,654 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 107,571 | 108,853 | 1,283 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 154,181 | 155,180 | 1,000 | All ST45 genomes | 100.00 | 99.60 | Lorraine | Homology to ISLpn9 |
| 172,917 | 184,420 | 11,504 | Unique to ZIB genomes | 80.00 | 89.22 | Present at much higher coverage | |
| 197,488 | 203,527 | 6,040 | Unique to ZIB genomes | 100.00 | 98.24 | Present at much higher coverage | |
| 203,528 | 228,760 | 25,233 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 254,469 | 255,506 | 1,038 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 258,026 | 259,597 | 1,572 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 261,477 | 263,700 | 2,224 | All ST45 genomes | 100.00 | 99.69 | Lorraine | Homology to ISLpn9 |
| 310,472 | 311,473 | 1,002 | Very low coverage in Philadelphia reference | 100.00 | 99.80 | Lorraine | Homology to ISLpn9 |
| 333,355 | 336,147 | 2,793 | All ST45 genomes | 100.00 | 99.96 | Lorraine Q958210.1 | |
| 359,496 | 360,497 | 1,002 | All ST45 genomes | 100.00 | 99.90 | Lorraine | Homology to ISLpn9 |
| 364,046 | 366,198 | 2,153 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 388,083 | 390,250 | 2,168 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 392,528 | 393,528 | 1,001 | All ST45 genomes | 100.00 | 99.80 | Lorraine | Homology to ISLpn9 |
| 470,948 | 471,949 | 1,002 | Very low coverage in Philadelphia reference | 100.00 | 99.90 | Lorraine | Homology to ISLpn9 |
| 492,613 | 493,614 | 1,002 | All ST45 genomes | 100.00 | 99.80 | Lorraine | Homology to ISLpn9 |
| 523,278 | 524,733 | 1,456 | Very low coverage in Philadelphia reference | 100.00 | 99.93 | Lorraine Q958210.1 | Homology to ISLpn9 |
| 578,328 | 579,663 | 1,336 | All ST45 genomes | 100.00 | 100.00 | NCTC11985 | |
| 620,762 | 622,020 | 1,259 | All ST45 genomes | 100.00 | 100.00 | NCTC11985 | |
| 622,674 | 660,984 | 38,311 | All ST45 genomes | 75.00 | 98.01 | Corby | |
| 698,965 | 699,965 | 1,001 | All ST45 genomes | 100.00 | 99.80 | NCTC11985 | Homology to ISLpn9 |
| 743,322 | 746,237 | 2,916 | All ST45 genomes | 100.00 | 100.00 | NCTC11985 | |
| 782,582 | 791,077 | 8,496 | All ST45 genomes | 100.00 | 99.80 | NCTC11985 | Across toxin gene |
| 795,792 | 798,713 | 2,922 | All ST45 genomes | 100.00 | 100.00 | NCTC11985 | |
| 812,799 | 818,434 | 5,636 | All ST45 genomes | 100.00 | 100.00 | NCTC11985 | |
| 925,770 | 927,658 | 1,889 | All ST45 genomes | 99.00 | 97.78 | ERS1305867 | |
| 944,897 | 946,813 | 1,917 | All ST45 genomes | 100.00 | 98.44 | Lorraine | |
| 1,048,512 | 1,050,216 | 1,705 | All ST45 genomes | 100.00 | 97.65 | ERS1305867 | |
| 1,059,149 | 1,060,776 | 1,628 | Very low coverage in Philadelphia reference | 100.00 | 99.63 | PATHC002 | Homology to ISLpn9 |
| 1,165,326 | 1,166,952 | 1,627 | Very low coverage in Philadelphia reference | 100.00 | 99.82 | Lorraine | Homology to ISLpn9 |
| 1,181,211 | 1,182,982 | 1,772 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 1,193,911 | 1,195,503 | 1,593 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 1,202,863 | 1,205,380 | 2,518 | All ST45 genomes | 100.00 | 99.80 | Lorraine | Homology to ISLpn9 |
| 1,227,451 | 1,231,629 | 4,179 | All ST45 genomes | 100.00 | 99.88 | Lorraine | |
| 1,233,358 | 1,235,843 | 2,486 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 1,237,708 | 1,239,143 | 1,436 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 1,333,023 | 1,337,543 | 4,521 | All ST45 genomes | 100.00 | 99.93 | Lorraine | |
| 1,355,443 | 1,356,680 | 1,238 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 1,358,599 | 1,360,228 | 1,630 | Very low coverage in Philadelphia reference | 100.00 | 99.94 | Lorraine | Homology to ISLpn9 |
| 1,439,798 | 1,441,093 | 1,296 | Very low coverage in Philadelphia reference | 100.00 | 99.46 | PATHC002 | Homology to ISLpn9 |
| 1,477,327 | 1,479,726 | 2,400 | All ST45 genomes | 100.00 | 99.62 | PATHC002 | Homology to ISLpn9 |
| 1,593,111 | 1,594,428 | 1,318 | Very low coverage in Philadelphia reference | 100.00 | 99.85 | Lorraine | Homology to ISLpn9 |
| 1,595,609 | 1,597,261 | 1,653 | All ST45 genomes | 100.00 | 100.00 | Lorraine | Lower homology to ISLpn9 |
| 1,605,942 | 1,606,940 | 999 | All ST45 genomes | 100.00 | 100.00 | Lorraine | Homology to ISLpn9 |
| 1,627,254 | 1,629,188 | 1,935 | All ST45 genomes | 100.00 | 100.00 | NCTC11985 | |
| 1,671,268 | 1,677,430 | 6,163 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 1,729,636 | 1,732,750 | 3,115 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 1,740,199 | 1,741,299 | 1,101 | All ST45 genomes | 100.00 | 99.64 | NCTC11985 | Homology to ISLpn9 |
| 1,750,367 | 1,752,008 | 1,642 | Very low coverage in Philadelphia reference | 100.00 | 99.70 | Lorraine | Homology to ISLpn9 |
| 1,778,688 | 1,781,029 | 2,342 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 1,869,245 | 1,871,275 | 2,031 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 1,896,804 | 1,900,876 | 4,073 | Very low coverage in Philadelphia reference | 100.00 | 99.80 | Lorraine | Homology to ISLpn9 |
| 1,971,818 | 1,972,865 | 1,048 | Very low coverage in Philadelphia reference | 100.00 | 99.90 | Lorraine | Homology to ISLpn9 |
| 2,017,691 | 2,018,953 | 1,263 | All ST45 genomes | 100.00 | 100.00 | Lorraine | Homology to ISLdr1 |
| 2,061,021 | 2,062,670 | 1,650 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,067,226 | 2,070,972 | 3,747 | Very low coverage in Philadelphia reference | 100.00 | 100.00 | Lorraine | Homology to ISLpn9 |
| 2,080,183 | 2,081,962 | 1,780 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,100,255 | 2,102,480 | 2,226 | Very low coverage in Philadelphia reference | 100.00 | 99.91 | Lorraine | Homology to ISLpn9 |
| 2,127,224 | 2,133,200 | 5,977 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,147,413 | 2,150,629 | 3,217 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,152,162 | 2,153,520 | 1,359 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,178,245 | 2,180,163 | 1,919 | Very low coverage in Philadelphia reference | 100.00 | 99.79 | Lorraine | Homology to ISLpn9 |
| 2,190,825 | 2,197,681 | 6,857 | All ST45 genomes | 100.00 | 99.97 | PATHC002 | |
| 2,216,894 | 2,217,895 | 1,002 | Very low coverage in Philadelphia reference | 100.00 | 99.90 | PATHC002 | Homology to ISLpn9 |
| 2,266,139 | 2,267,687 | 1,549 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,286,060 | 2,289,391 | 3,332 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,292,710 | 2,314,659 | 21,950 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,315,009 | 2,316,667 | 1,659 | Very low coverage in Philadelphia reference | 100.00 | 100.00 | Lorraine | |
| 2,319,117 | 2,324,373 | 5,257 | All ST45 genomes | 100.00 | 1005.00 | ||
| 2,327,309 | 2,331,362 | 4,054 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,367,178 | 2,368,459 | 1,282 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,374,126 | 2,376,841 | 2,716 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,395,108 | 2,396,866 | 1,759 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,398,121 | 2,399,129 | 1,009 | Very low coverage in Philadelphia reference | 100.00 | 99.70 | Lorraine | Homology to ISLpn9 |
| 2,437,537 | 2,438,802 | 1,266 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,530,688 | 2,531,689 | 1,002 | Very low coverage in Philadelphia reference | 100.00 | 99.80 | Lorraine | Homology to ISLpn9 |
| 2,562,295 | 2,563,296 | 1,002 | All ST45 genomes | 100.00 | 100.00 | NCTC11985 | Homology to ISLpn9 |
| 2,624,142 | 2,697,268 | 73,127 | All ST45 genomes | 100.00 | 99.99 | Lorraine | |
| 2,700,063 | 2,708,857 | 8,795 | All ST45 genomes | 100.00 | 99.73 | Lorraine | |
| 2,711,396 | 2,713,990 | 2,595 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,720,705 | 2,728,751 | 8,047 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,755,536 | 2,756,800 | 1,265 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,760,534 | 2,762,270 | 1,737 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,774,564 | 2,776,814 | 2,251 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,784,580 | 2,788,461 | 3,882 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,871,046 | 2,873,896 | 2,851 | Very low coverage in Philadelphia reference | 100.00 | 100.00 | PATHC002 | Homology to ISLpn9 |
| 2,877,959 | 2,880,331 | 2,373 | All ST45 genomes | 100.00 | 99.96 | Lorraine | |
| 2,901,276 | 2,903,059 | 1,784 | Very low coverage in Philadelphia reference | 100.00 | 99.94 | Lorraine | Homology to ISLpn9 |
| 2,927,856 | 2,930,694 | 2,839 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,938,791 | 2,941,692 | 2,902 | All ST45 genomes | 100.00 | 99.97 | Lorraine | Homology to ISLpn11 |
| 2,944,637 | 2,949,035 | 4,399 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,949,872 | 2,951,925 | 2,054 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,967,128 | 2,974,009 | 6,882 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,980,896 | 2,986,006 | 5,111 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,988,838 | 2,993,936 | 5,099 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 2,996,660 | 3,000,260 | 3,601 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 3,002,511 | 3,004,154 | 1,644 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 3,022,889 | 3,025,113 | 2,225 | All ST45 genomes | 100.00 | 100.00 | PATHC002 | Homology to ISLpn9 |
| 3,135,943 | 3,137,185 | 1,243 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 3,210,927 | 3,212,090 | 1,164 | Very low coverage in Philadelphia reference | 100.00 | 98.63 | Lorraine | Homology to ISLpn9 |
| 3,322,005 | 3,323,006 | 1,002 | Very low coverage in Philadelphia reference | 100.00 | 99.90 | Lorraine | Homology to ISLpn9 |
| 3,380,474 | 3,381,474 | 1,001 | Very low coverage in Philadelphia reference | 100.00 | 99.70 | Lorraine | Homology to ISLpn9 |
| 3,395,571 | 3,398,446 | 2,876 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 3,405,473 | 3,406,634 | 1,162 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 3,411,700 | 3,413,038 | 1,339 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 3,413,490 | 3,416,746 | 3,257 | All ST45 genomes | 100.00 | 100.00 | Lorraine | |
| 3,462,908 | 3,464,229 | 1,322 | All ST45 genomes | 100.00 | 99.62 | Lorraine | Homology to ISLpn9 |
| 3,494,277 | 3,495,904 | 1,628 | Very low coverage in Philadelphia reference | 100.00 | 99.63 | PATHC002 | Homology to ISLpn9 |
| 3,520,489 | 3,521,921 | 1,433 | All ST45 genomes | 94.00 | 99.93 | H3 P114576.1 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLegionella and Acanthamoeba research · Water Treatment and Disinfection · Bacterial biofilms and quorum sensing
Data Summary
All data are submitted to the ENA under project PRJEB79004 under accession nos. ERR13662450–ERR13662572 (Table S1). Supplementary material is available on Figshare: https://doi.org/10.6084/m9.figshare.28387712.v1 [1].
Introduction
Legionella pneumophila, a Gram-negative, aerobic and non-spore-forming bacterium, is an opportunistic pathogen that can present a risk to human health. It can cause Legionnaires’ disease, a severe atypical pneumonia, or Pontiac fever, a milder form of the same infection [2]. Of the over 60 known Legionella species, L. pneumophila serogroup 1 is responsible for most severe infections. Transmission from environment to human occurs through inhalation or aspiration of aerosols containing L. pneumophila, where the bacteria infect alveolar macrophages in the lung [34].
L. pneumophila are ubiquitous in various freshwater habitats as well as man-made water systems such as potable water systems, fountains, spa water and air-conditioner cooling towers [25]. In these environments, L. pneumophila can be found planktonically or coexisting with other micro-organisms in biofilms where they can parasitize and replicate within free-living amoebae [6]. The association with amoebae offers benefits to Legionella, providing protection against disinfectants, UV radiation or fluctuations in water temperature [7]. Water temperatures between 25 and 45 °C are optimum for growth, at which L. pneumophila may reach critical concentrations. At higher water temperatures, bacterial growth is inhibited, and temperatures above 60 °C start to be bactericidal for L. pneumophila.
Due to this ability to grow within pipes, guidelines and regulations exist to control the growth of Legionella spp. in the water systems of buildings. In addition to sanitary measures such as regular maintenance of the waterlines or temperature checks, regular microbiological analysis of water samples is recommended to monitor the water quality [89]. This also applies to healthcare facilities, such as hospitals or clinics, which may treat vulnerable persons having a potential higher risk of acquiring Legionnaire’s disease. In dental institutions, biofilm formation in dental unit water lines may result in high numbers of micro-organisms including Legionella spp. in the water used for cooling or ultrasonication procedures [10]. Consequently, an increased risk for Legionella infection has also been assumed for dental health care workers; however, a recent review suggests that there is only very limited evidence for an increased risk in this professional group [11].
Epidemiological studies based on the analysis of genomic data are very helpful in identifying transmission routes and tracing potential sources of infections. In addition, such studies can also be useful to assess the microbial diversity and evolutionary relatedness in various environments. Genomic epidemiological analysis, together with aggressive interventional measures, was central to controlling an outbreak of nosocomial cases of L. pneumonia in an Australian hospital [12]. In this case, eradication methods, which included disinfection of the water distribution system with a chlorinated, alkaline detergent as well as removal of redundant plumbing systems, appeared to be successful in eliminating the L. pneumophila population responsible for the infections in the hospital for at least six months. However, other studies show that artificial water systems remain colonized by L. pneumophila despite the implementation of decontamination measures such as the superheat and flush method [1314].
At the Dental School of the University of Basel, Switzerland, routine water testing began in the early 1990s with a focus on tap water to assess and control bacterial contamination at the source level before it could enter the waterlines of dental units. In this context, a unique collection of L. pneumophila strains isolated from different points in the water system was created, before and after decontamination measures, in the same water distribution system, from 1994 to 2018. In summer 2019, the Dental School of the University of Basel moved to a new building in a different location.
The aim of this study was to investigate the distribution and evolution of L. pneumophila within a single building over 25 years.
Methods
Study setting and sample collection
Routine testing of the drinking water system was performed annually at the Dental School of the University of Basel, Switzerland, on a regular basis. The building has housed the Dental School of the University of Basel for almost one hundred years and was, where necessary, modified to meet new requirements during this period. In particular, the historic part of the building contained older water pipes and was not initially designed to prevent potential colonization by Legionella spp.
Therefore, the sampling points were selected based on a risk assessment and included end points of the water distribution system as well as rarely used faucets. Additional tests were carried out as required and after consultation with the Hygiene Commission of the building. Water samples were usually taken from the warm water circuit and processed in accordance with national guidelines that were in force at the respective time [9]. Following species identification, L. pneumophila isolates were stored at −70 °C until analysis. As required by the guidelines, 200 ml of water was taken from the respective water outlet and filtered by pressure through a membrane filter. The filter was then placed onto an agar plate selective for Legionella species. Prior to 2008, Legionella MWY selective medium (Oxoid, Pratteln, Switzerland) was used, whereas from 2008 on, GVPC medium (Oxoid, Thermo Scientific, Reinach, Switzerland) was used for the initial isolation of Legionella spp. from the water samples. When reporting the total c.f.u., numbers are calculated to 1 l of water.
Culture and DNA extraction
L. pneumophila strains isolated during the routine water tests in the years 1994 to 2018, i.e. during a period of 25 years, were included in this study. The isolates were grown on buffered charcoal yeast extract agar (Thermo Fisher Scientific, Reinach, Switzerland) aerobically in a humidified atmosphere at 37 °C for 48–72 h. DNA was extracted using the Qiagen EZ1 with the DNA tissue kit (Qiagen AG, Hombrechtikon, Switzerland).
Genome sequencing and analysis
All samples were sequenced on the NextSeq500 PE150 after Illumina DNA prep, resulting in over 30× mean read depth per sample. Isolate ZIB8567 was sequenced by Oxford Nanopore Technologies on an R10.4 flowcell to a mean read depth of 50× and base called with Dorado [email protected] (https://github.com/nanoporetech/dorado). All data were submitted to the European Nucleotide Archive (ENA) under project PRJEB79004.
Ridom v 9.0.10 was used on Unicycler v0.3.0b [15] assemblies to assign sequence-based typing sequence types (STs) to the isolates. Data on two ST45 isolates (917,837 and 919,569) were obtained from the UK Health Security Agency (UKHSA) and used as outgroups.
For phylogenetic analysis, the hybrid Unicycler v0.4.8 [15] assembled genome of isolate ZIB8567 (isolated in 2018), comprising a single contig of 3,522,222 bp (accession OZ182546.1), was used as the reference against which to map the reads of all isolates within CLC Genomics Workbench v22.0.2, calling variants with parameters: variant calling with 10× minimum coverage, 10 minimum count and 70% minimum frequency. Consensus genomes were extracted from mapped reads using a minimum coverage of 10. A multiple sequence alignment was created from the whole genome alignments, exported and run through Gubbins v3.3.0 [16] with five iterations to remove recombinations. Results were viewed in Phandango [17]. BactDating v1.1.1 [18] was run four times on the Gubbins output with one million iterations and the arc model to check the reproducibility, and representative results are given. Abricate v1.0.1 (https://github.com/tseemann/abricate) and the NCBI database [19] were used to define resistance determinants present.
Regions of the genome present in the sequenced genomes and absent from the genomes of strain Philadelphia (SRR801743 and ERR351242), or present at very low coverage, were identified by analysing the mapping statistics of all isolates in CLC (<99.9% of reference mapped to), visualizing mapped reads in Artemis and marking regions without mapping. Regions over 1 kb were compared with blastn against the nt database to check whether there were matches in other submitted genomes, and top matches are given. Insertion elements (ISs) were categorized using ISFinder (https://isfinder.biotoul.fr). Further elements were characterized by iterative analysis of the mapping statistics (<98% reads mapped) and de novo assembly of Illumina data in CLC using default parameters (slow) and mapping of reads as above. Loss and gain of elements were confirmed using Panaroo v1.3.0 [20] on assemblies created with Unicycler v0.4.8 [15] annotated with Bakta v1.9.3 [21] within the IMMense pipeline (https://gitlab.uzh.ch/appliedmicrobiologyresearch/immense), and assessing resulting gene presence/absence files in Phandango [17].
Results
Sampling
Overall, 309 water samples, taken from different locations in the building, were collected at 38 time points over a period of 25 years. As the building, including waterlines, was renovated over the course of the 25 years at various locations, the same sampling points might not have been accessible any longer, and therefore, nearby faucets fed by the same waterline were selected. L. pneumophila was recovered from 120 water samples (38.8%) and 113 L. pneumophila isolates from 26 time points could be included in this study (Fig. 1), as well as 10 replicate isolates picked from the same plates as 5 of the isolates. After initial control measures, which to this date consisted mainly of two cycles of heat decontamination and the installation of a new boiler system, which were successful for ~11 years until 2007, an increase in the total number of Legionella c.f.u. as well as of L. pneumophila-positive sites was noticed (Fig. 1).
Timeline of L. pneumophila strains isolated over 25 years. Shown are the number of sites tested per time point, the number of sites that were L. pneumophila positive, the minimum and maximum c.f.u. l−1 values for L. pneumophila-positive sites per time point and the number of isolates per time point that was analysed (in bold) as well as the decontamination measures carried out during this time period.
Phylogenetic analysis of ST45 cluster
All 123 isolates from the building belong to ST45, which is within serogroup 1. They are unrelated to previously published samples from other locations within the City of Basel, where the Dental School of the University of Basel is located [22].
Phylogenetic analysis of the 123 isolates, based on a complete genome hybrid assembly reference from an isolate within the cluster, shows that they are closely related (Fig. 2). We found 408 SNPs across the whole cluster, with recombinations removed. There is no apparent clustering of isolates taken from the same sampling points. Similarly, there is no clear clustering between replicate colonies which were taken from some agar plates in 1997, 2009 and 2018, with samples from the same plates being found at diverse locations in the tree (Fig. 2). The branch which particularly expanded post-2008 (Fig. 2) carries a C-A SNP at position 377 of the transfer-messenger RNA (tm-RNA) ssrA.
Recombination-adjusted phylogenetic tree of all isolates with associated metadata. Gubbins generated a phylogeny based on mapping all data against a complete hybrid assembly of ZIB8567, rooted on the oldest isolate (ZIB9001, 1994). Metadata (right) shows (left to right) which replicate isolates came from the same plates (replicate colonies), year of isolation (date) and location of isolation within the building (place). As some of the same sampling points were not always accessible during the 25 years, nearby faucets fed by the same waterline were selected and/or additional water outlets were included. Water outlets fed by the same waterline are represented by the same number. Figure generated using Phandango [17]. The branch shown in red carries samples isolated exclusively after 2008.
A mutation rate of 0.38 SNPs per genome per year (range 0.27–0.49) and a most likely date for the common ancestor of this cluster of 1938 (range 1911–1959) were calculated using BactDating. The two ST45 isolates submitted to the UKHSA originating from Oxford in 2009 (917,837) and of unknown provenance (919,569) are, respectively, 1,875 and 1,888 SNPs from the ST45 isolates sequenced for this study (recombinations removed). The recombinations identified within the cluster represent 94% of the identified SNPs (6,876/7,284), although many fall within repeat genes and may represent mapping difficulties (Fig. S1, available in the online Supplementary Material).
Regarding resistance determinants, the gene aph(9)-Ia, a chromosomally encoded aminoglycoside phosphotransferase, was identified in the hybrid-assembled genome of L. pneumophila ZIB8567 with a nucleotide identity of 89.81%.
Regions of difference and genomic islands
All the presented L. pneumophila genomes share very similar gene content, with reads from all genomes mapping to over 99% of the ZIB8567 hybrid assembly. This is also the case for the ST45 genomes 917,837 and 919,569 obtained from UKHSA. Reads from the L. pneumophila strain Philadelphia, however, cover only 87% of the assembly, with under 90% of the reads mapping.
Many regions of difference (RoD) were identified in the ST45 genomes which are not present in the Philadelphia reference strain genome (Table 1). When comparing the two ST45 database genomes, only one possible genomic island (GI) appears to be unique to the isolates from this study: a 30 kb region (172,917–203,527 in the ZIB8567 hybrid assembly) adjacent to the tm-RNA ssrA, sharing 90.55% identity over 92% of its length with a region of the genome of Legionella longbeachae strain B1445CHC (CP045306.1). This RoD shows a higher read depth than the rest of the genome, suggesting that it exists in multi-copy and carries phage-like genes with a type IV secretion system. This RoD is absent from the genomes of six isolates, namely ZIB8547, ZIB9003, ZIB9004, ZIB9008, ZIB9009 and ZIB9016, all originating from close sampling points 2A and 2B. The position of these isolates in the phylogeny infers that the ancestral strain of this cluster carried the RoD, and it had been lost on three independent occasions within the phylogeny.
All the rest of the RoD between ST45 and Philadelphia are present in the genomes of other strains of L. pneumophila, with most matches to that of strain Lorraine (FQ958210.1). Forty of the RoDs appear to be IS elements, of which the vast majority (n=38) are related to ISLpn9 (Table 1).
The isolates ZIB8557, ZIB9043, ZIB9046, ZIB9051, ZIB9053, ZIB9054, ZIB9063, ZIB9064 and ZIB9072, all from locations 4A and 4B, carry a 150 kb plasmid identical to pB3526CGC_150 k from L. longbeachae (CP042253.1). This plasmid carries blaOXA-29, among other potential antimicrobial resistance encoding genes and those potentially encoding conjugation apparatus (Files S1 and S2). According to the phylogeny, this suggests acquisition of the plasmid on three separate occasions.
Discussion
L. pneumophila has been reported from the water systems of large buildings and hospitals in studies from Canada [23], the USA [132425], Italy [26], Germany [27], Finland [14], Poland [28], Scotland [29] and Australia [12]. Genome sequencing has previously been used in such studies [123031], but none over such a long period of time as in our study.
Our data show a long-term presence of L. pneumophila ST45 within an old building in Basel in which the Dental School of the University of Basel was located from 1924 to 2019. The estimated time of the most recent common ancestor of the cluster, at around 1938, would therefore fit with the age and development of the building, as this historical building has been regularly renovated and new annexes added [32]. While some buildings have been found to contain multiple STs [31], our study describes the same ST found in all locations at all time points, suggesting continuous colonization of the waterlines with the same clonal strain over 25 years. That there was no clear phylogenetic signal related to location within the building infers circulation through the waterlines and less spatial structuring than described by David et al. [31]. Likewise, no influence of the decontamination measures could be noted in the phylogeny in terms of the extinction of certain branches. The increase in prevalence observed from 2007 could partly reflect a change in the Standard Operating Protocol (SOP) to use a different selective agar medium, although isolates indeed grew on the first medium from 1994 to 2007. Because of this increase, heat decontamination was carried out in 2009, but this did not lead to a reduction in the culture of Legionella spp. One SNP of interest is that in ssrA, which is also adjacent to a possible mobile GI; inhibition of expression of this tm-RNA [33] has severely deleterious effects on bacterial growth [34]. It is possible that a more in-depth analysis including more isolates from each time point might have resulted in a more nuanced picture in terms of clonal extinction, as decontamination measures have been shown to reduce the number of Legionella present [3536]. This might also have helped us to capture the presence of any L. longbeachae isolates.
Investigation of global data provided by UKHSA and from the literature shows that L. pneumophila ST45 has previously been isolated in environmental samples from China, Japan, South Korea, France, Germany, Belgium and Italy, and in clinical isolates (n=41) from the USA, Canada and across Europe, including from two locations in Switzerland and the neighbouring state in Germany (Baden-Württemberg) between 2010 and 2013 [3742]. Very few genomes are available for comparison, and the two ST45 genomes available were over 1,800 SNPs away from the presented cluster. Isolates within this ST have been found to cause clinical disease, yet no infections were identified within our study. It is commonly assumed that dental personnel have an elevated occupational risk for Legionella infection, as they might potentially inhale contaminated aerosols generated by water-cooled rotary instruments used in dentistry [43]. However, no cases of Legionella infection were reported over the course of our study period among dental personnel with direct patient contact working at the Dental School of the University of Basel (data not shown). This is in accordance with a recent meta-analysis that concluded that the occupational risk for an occupationally acquired Legionella infection for dental personnel is lower than previously anticipated [11] and strict compliance with currently valid guidelines may have contributed to the absence of Legionella infections among dental personnel at the Dental School of the University of Basel. Nevertheless, awareness, regular testing and other control measures, e.g. the use of sterile water for critical procedures, are recommended.
The calculated mutation rate within this cohort of 0.38 SNPs per genome per year compares well to previous estimates of 0.71 SNPs per genome per year (44) and 0.49 SNPs per genome per year (45). L. pneumophila is known to be a recombinogenic species [4647]; the present study shows limited recombination, as is likely in an environment with limited genomic diversity, analysis of which may be confounded by repetitive genes. Recombinations around hemA, identified as a hotspot in ST1 [47], were not identified in this ST45 cluster. Plasmid movement and GI loss were also observed, showing that genome plasticity is possible in this population. Interestingly, these events are associated with isolates from particular locations; however, it is not possible to infer specific selective pressure on these elements within these niches. Genetic elements of * L. longbeachae* could be detected, although this bacterium itself was not isolated in this study. Other Legionella species, such as Legionella geestiana and Legionella anisa, were detected, albeit rarely (data not shown).
In conclusion, the present work demonstrates that a single clonal lineage of L. pneumophila can, despite decontamination measures, persist in a building’s water system for over 25 years. The phylogeny of the isolates can be interpreted as inferring good water circulation and possible recolonization from a common source after cleaning.
Supplementary material
10.1099/mgen.0.001393Uncited Table S1.
10.1099/mgen.0.001393Uncited Fig. S1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Seth-Smith HM Egli A Gautsch S Bornstein MM Kulik EM Genomic analysis of Legionella pneumophila in the drinking water system of a large building over 25 years Microbiology Society. Dataset 202510.6084/m 9.figshare.28387712.v 1PMC 1228230040408137 · doi ↗ · pubmed ↗
- 2Fields BS Benson RF Besser RE Legionella and Legionnaires’ disease: 25 years of investigation Clin Microbiol Rev 20021550652610.1128/CMR.15.3.506-526.200212097254 PMC 118082 · doi ↗ · pubmed ↗
- 3Gao LY Harb OS Abu Kwaik Y Utilization of similar mechanisms by Legionella pneumophila to parasitize two evolutionarily distant host cells, mammalian macrophages and protozoa Infect Immun 1997654738474610.1128/iai.65.11.4738-4746.19979353059 PMC 175680 · doi ↗ · pubmed ↗
- 4Chauhan D Shames SR Pathogenicity and virulence of Legionella: intracellular replication and host response Virulence 2021121122114410.1080/21505594.2021.190319933843434 PMC 8043192 · doi ↗ · pubmed ↗
- 5Wuthrich D Gautsch S Spieler-Denz R Dubuis O Gaia V et al Air-conditioner cooling towers as complex reservoirs and continuous source of legionella pneumophila infection evidenced by a genomic analysis study in 2017, switzerland. euro surveillance: bulletin europeen sur les maladies transmissibles = european communicable disease bulletin Euro Surveill 20192410.2807/1560-7917.ES.2019.24.4.1800192 PMC 635199430696527 · doi ↗ · pubmed ↗
- 6Hilbi H Buchrieser C Microbe Profile: Legionella pneumophila - a copycat eukaryote: this article is part of the microbe profiles collection Microbiology [Internet]2022 Mar 1https://www.microbiologyresearch.org/content/journal/micro/10.1099/mic.0.001142 accessed 21-October-202410.1099/mic.0.00114235230931 · doi ↗ · pubmed ↗
- 7Molmeret M Horn M Wagner M Santic M Abu Kwaik Y Amoebae as training grounds for intracellular bacterial pathogens Appl Environ Microbiol 200571202810.1128/AEM.71.1.20-28.200515640165 PMC 544274 · doi ↗ · pubmed ↗
- 8Van Kenhove E Dinne K Janssens A Laverge J Overview and comparison of Legionella regulations worldwide Am J Infect Control 20194796897810.1016/j.ajic.2018.10.00630638676 · doi ↗ · pubmed ↗