Total Syntheses of (±)-Lepadiformine B and C
Wei-Ting Hsiao, Jui-Lin Wu, Wen-Hua Chiou

TL;DR
This paper describes the complete chemical synthesis of two compounds, lepadiformine B and C, using a stereodivergent approach.
Contribution
The novel contribution is a stereodivergent synthesis method for N-acetyl-2-alkyl-8a-cyanodecahydroquinoline derivatives.
Findings
A deprotection-initiated alkylative/reductive cyclization method was developed for stereodivergent synthesis.
Lepadiformine B and analogs were synthesized via Ir-catalyzed reductive cyanation and other reactions.
The method allows for the preparation of both cis- and trans- isomers efficiently.
Abstract
Total syntheses of (±)-lepadiformine B and C are presented. The key theme in our approach is the stereodivergent synthesis of both cis- and trans- N-acetyl-2-alkyl-8a-cyanodecahydroquinoline, which are effectively prepared through deprotection-initiated alkylative/reductive cyclization of sterically well-defined α-aminonitriles bearing a masked carbonyl group. After the Dieckmann-type condensation to a tricyclic lactam, lepadiformine B and its analogues can be achieved through the Ir-catalyzed reductive cyanation and subsequent hydrolysis, reduction, and Bruylants reactions.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
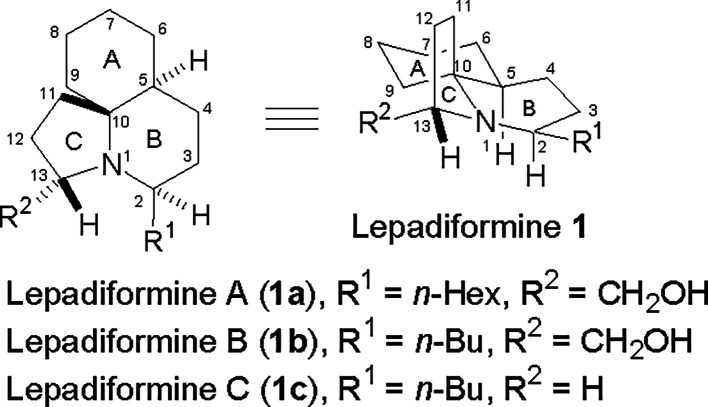 Figure 1
Figure 1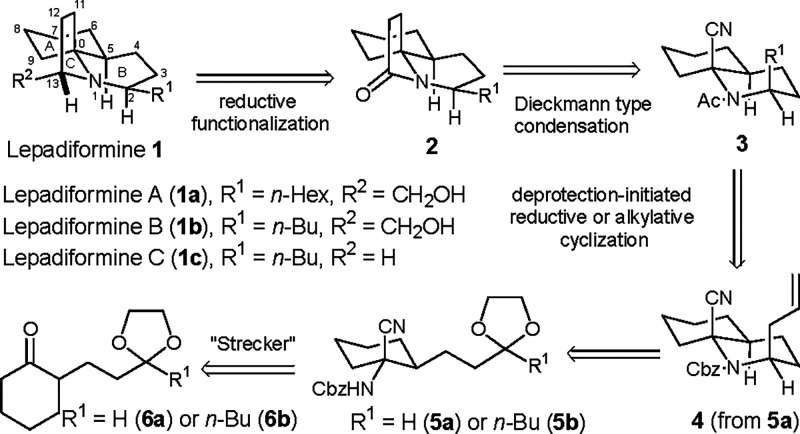 Figure 2
Figure 2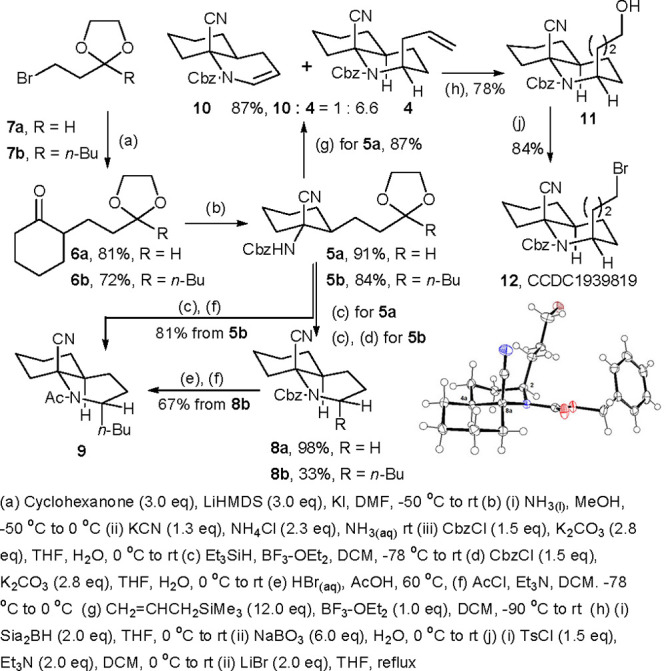 Figure 3
Figure 3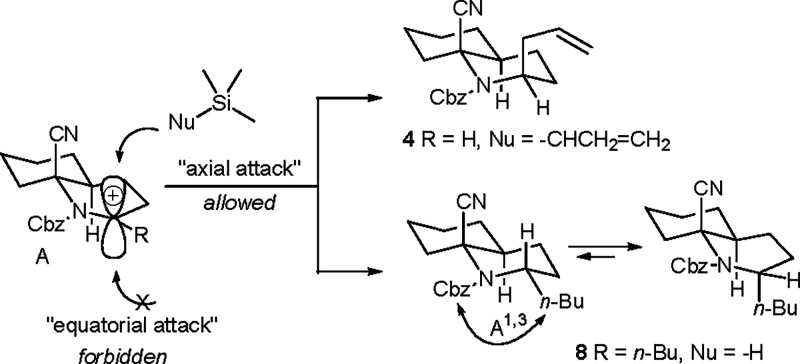 Figure 4
Figure 4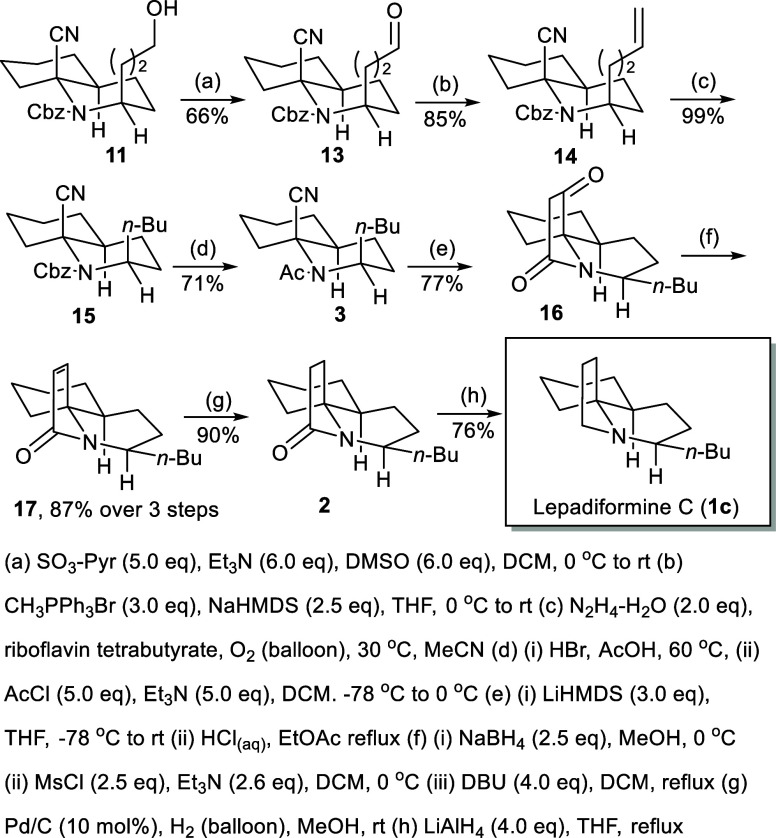 Figure 5
Figure 5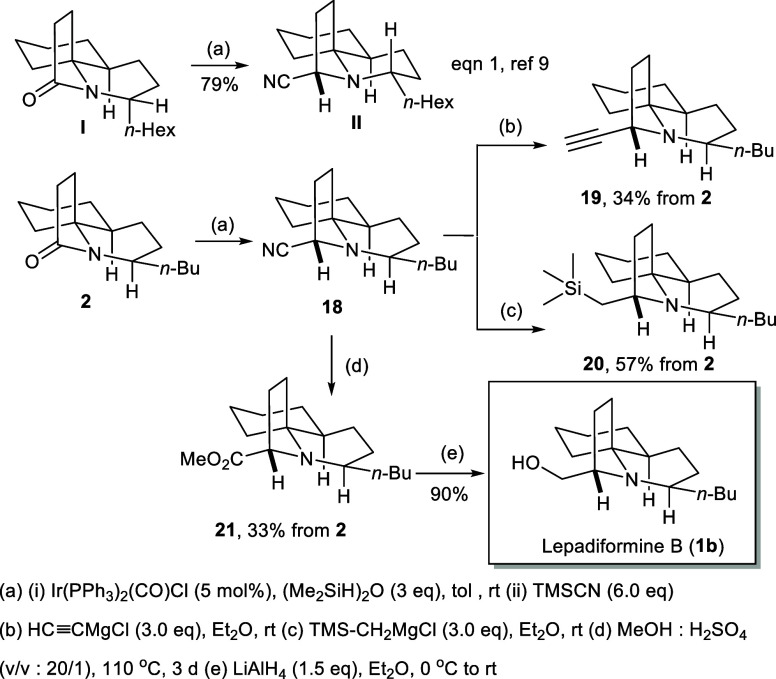 Figure 6
Figure 6 Figure 7
Figure 7 Figure 8
Figure 8- —National Science Council10.13039/501100001868
- —National Science Council10.13039/501100001868
- —National Science Council10.13039/501100001868
- —National Science Council10.13039/501100001868
- —National Science Council10.13039/501100001868
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced Synthetic Organic Chemistry · Chemical Synthesis and Analysis · Chemical synthesis and alkaloids
In 2006, Sauviat et al. reported the isolation of marine alkaloids lepadiformine B (1b) and lepadiformine C (1c) (Figure) from the tunicate Clavelina moluccensis Sluiter collected in Djibouti waters.? Lepadiformine B (1b) and C (1c) are tricyclic alkaloids that possess a tricyclic perhydropyrido[2,1-j]-quinolone structure bearing an n-butyl group at the C2 position, and their difference is the presence of the hydroxymethyl group at the C13 position in 1b. Lepadiformine B (1b) shows moderate inhibition of the cardiac inward rectifier K^+^ channel compared to its analogue lepadiformine A (1a) bearing an n-hexyl group, while 1c shows poor inhibition activity. These findings suggest that the length of the C2 side chain and the presence of the C13 hydroxymethyl group do matter. In addition, these lepadiformine alkaloids also exhibit moderate cytotoxicity against several tumor cell lines.? As the first reported member in the lepadiformine family, lepadiformine A (1a) was isolated by Biard in 1994, from the marine ascidian Müller in Tunisia.? The correct structure was confirmed through total synthesis by Kibayashi and co-workers in 2000.? The interesting structure and biological activities have prompted chemists to develop various intellectual and elegant strategies, simply classified as racemate syntheses,? enantiomeric syntheses, ?,? and reviews.?
Recently, we developed a divergent approach to both cis- and trans-N-acetyl-2-alkyl-8a-cyano-decahydroquinoline (8a-CDHQ) structures via a double consecutive epimerization from the same intermediate Cbz-protected cis-2-allyl-8a-CDHQ. Subsequent base-mediated Dieckmann-type condensation of the N-acetyl-2-alkyl-8a-CDHQs furnished a tricyclic lactam with either cis or trans configuration in the B ring.? Through this strategy, we completed the synthesis of fasicularin, a tunicate alkaloid.? In this article, we describe the extension of this work, intended for use in the total syntheses of (±)-lepadiformine B (1b) and C (1c).
Our retrosynthetic analysis is shown in Scheme. The hydroxymethyl group at C13 can be introduced by a one-carbon elongation of tertiary lactam 2 through Dixon’s reductive cyanation? and subsequent transformations. A Dieckmann-type condensation between the angular nitrile with the acetyl group in cis-acetamide 3 can generate the pyrrolidine C ring of lactam 2. Different from the approach to fasicularin, in which the necessary trans configuration in the B ring is achieved through epimerization, conversion to cis-acetamide 3 from the known Cbz-protected cis-2-allyl-8a-cyanodecahydroquinoline (4) involves relative configuration retention. Although cis-8a-CDHQ 4 can be available based on our previously established method, an improved procedure has been developed for more efficient preparation. If the α-aminonitrile derivative 5a bearing a propyl substituent with a masked aldehyde group, i.e., a terminal ethylene acetal, is reacted with Lewis acid and allyltrimethylsilane, spontaneous cyclization to 8a-CDHQ 4 may proceed in a domino manner: simultaneous cleavage of the dioxolane moiety and formation of a transient piperidinium, followed by addition with allyltrimethylsilane. Similarly, as an α-aminonitrile bearing an ethylene ketal group with the correct position and side chain length is reacted within Lewis acid and triethylsilane, it will result in the formation of 8a-CDHQ with the opposite configuration at the C2 position, as long as both additions follow the identical mechanism. Such a deprotection-initiated alkylative/reductive cyclization approach may save manipulations for the construction of the B ring and the side chain and provide divergent syntheses for both cis- and trans-8a-CDHQ. The sterically well-defined α-aminonitrile 5 can be obtained by a Strecker reaction of the α-monosubstituted cyclohexanone derivative 6.
Our synthesis commences with the preparation of 6 through the α-alkylation of cyclohexanone. To find the optimal α-alkylation conditions, commercially available 1-bromo-3,3-ethylenedioxypropane (7a) was chosen for the optimization of the conditions of alkylation based on Kita’s protocol? (see Supporting Information). Treatment of bromide 7a with 3 equiv of cyclohexanone and LiHMDS in DMF at −50 °C proceeded smoothly to give 81% yield of the desired product 6a (Scheme). The analogue 6b was also achieved in 72% isolated yield with bromide 7b. The subsequent Strecker reaction of 6a, i.e., with KCN in ammonia at room temperature, and CbzCl protection proceeded readily to deliver single α-aminonitrile 5a in 91% yield. This tactic was employed to obtain 5b in 84% yield.
The conditions for the deprotectively reductive cyclization were investigated (see Supporting Information for more details); we found that the treatment of α-aminonitrile 5a with 1.0 equiv of BF_3_·OEt_2_ and triethylsilane afforded the cyclized product 8a in excellent yield. The application of the conditions on aminonitrile 5b did not yield the expected cyclized product, but a Cbz-free uncyclized product, which was then subjected to protection with CbzCl back to the original 5b. Treatment with 1.3 equiv of BF_3_·OEt_2_ and triethylsilane resulted the formation of a Cbz-free cyclized product, which was protected with CbzCl to produce the cyclized product 8b in 33% yield. The results showed substantial reactivity difference between acetal 5a and ketal 5b. For acetal 5a, cleavage of the dioxolane group proceeds first, followed by reductive cyclization, while the removal of the Cbz group proceeds first for ketal 5b, followed by the cleavage of dioxolane and reductive cyclization. Such a series of reactions could be viewed as a domino process.? However, to our dismay, the NMR spectra of 8b were not identical to those of the known Cbz-protected cis-2-butyl-8a-CDHQ, and this might suggest a trans product. To verify the supposition, the Cbz group was transformed to an acetyl group using the protocol of HBr/AcOH deprotection and then acetylation. The spectra of the resulting product were consistent with those of known trans-N-acetyl-2-n-butyl-8a-CDHQ (9). Hence, the results confirm that the deprotectively reductive cyclization of 5b affords the product with the trans configuration rather than the desired cis configuration. Even so, as a crucial intermediate for the synthesis of fasicularin, the yield of the trans acetyl product 9 was improved up to 81% through direct treatment of 5b with 2 equiv BF_3_·OEt_2_ and triethylsilane followed by acetylation. This concise approach allows the rapid preparation of trans-9 within three steps from 7b through simple operations.
Since deprotectively reductive cyclization of ketal 5b affords the trans product, we may infer that deprotectively allylative cyclization of acetal 5a will result in the formation of the cis product. Thus, treatment of 5a with BF_3_·OEt_2_ and allyltrimethylsilane triggered a deprotection-initiated allylative cyclization and resulted in an inseparable mixture of the desired cis-2-allyl-8a-CDHQ (4) and enecarbamate 10 (∼6.6:1) in 87% combined yield (for optimization of the deprotection-initiated allylative cyclization, see Supporting Information). Subjecting the mixture to Sia_2_BH-mediated hydroboration/oxidation could yield primary alcohol 11 in 78% yield and enecarbamate 10 in 16% yield. The spectral data of the products were consistent with our previously published values. Moreover, to our delight, simple treatment of primary alcohol 11 with TsCl followed by substitution with LiBr gave a yield of 84% of crystalline bromide 12, whose structure was established unequivocally by X-ray analysis (CCDC No. 1939819), corroborating the present cis configuration in primary alcohol 11 (Scheme). Thus, we accomplished divergent syntheses for the rapid construction of both cis- and trans-2-alkyl 8a-CDHQs: a cis-8a-CDHQ, e.g., 4, could be achieved through BF_3_·OEt_2_-mediated deprotectively allylative cyclization of acetal 5a, while a trans-8a-CDHQ, such as 9, could be achieved through BF_3_·OEt_2_-mediated deprotectively reductive cyclization of ketal 5b.
The results could be rationalized by the Fürst–Plattner rule,? in which the nucleophile proceeds in an axial orientation to avoid a boat-like transition state (Scheme).? The conformation of the B ring in 8 should take a boat form to reduce the A ?,? strain of the carbamate from the n-butyl group, which has been observed in the X-ray structures of many analogues in our previous findings. ?,? The synthetic route provides a divergent route for the rapid construction of both cis and trans-2-alkyl CDHQs 4 and 9.
With alcohol 11 in hand, the n-butyl appendage could be achieved according to a three-step protocol (Scheme): the Parikh–Doering oxidation to aldehyde 13 in 66% yield, the Wittig olefination with Ph_3_PCH_2_ to olefin 14 in 85% yield, and flavin-mediated aerobic hydrogenation conditions to 15 in 99% yield.? Aerobic hydrogenation conditions only effect saturation of the alkene portion but do not remove the Cbz group, which avoids the double consecutive epimerization initiated by Pd-catalyzed hydrogenation conditions. The Cbz portion of 15 was removed within HBr in acetic acid followed by acetylation to furnish acetamide 3 in 71% overall yield. Treatment of acetamide 3 with LiHMDS followed by acidic workup afforded tricyclic ketone 16 in 77% yield. After the reduction of the ketone group of 16 with NaBH_4_, the resulting alcohol was mesylated and then eliminated by DBU to furnish conjugated lactam 17 in 87% yield over three steps. Catalytic hydrogenation at ambient pressure saturated the olefin to tricyclic lactam 2 in 90% yield. LiAlH_4_ reduction of lactam 2 afforded a 76% yield of lepadiformine C (1c) whose spectral properties were in accordance with the reported data.?
With access to the key tricyclic lactam 2, the introduction of a substituent on the γ-lactam could be addressed through Dixon’s Ir-catalyzed hydrosilylation–cyanation protocol (Scheme).? Lactam 2 was able to undergo reductive cyanation to provide aminonitrile 18, as demonstrated by our successful transformations in the synthesis of fasicularin (I to II, eq 1; Scheme). However, unlike tricyclic α-aminonitrile bearing a trans-B ring, the tricyclic *cis-*α-aminonitrile 18 appeared to be labile and easily oxidized back to lactam 2, especially after column chromatography or a longer standing. Therefore, we envisioned that the conversion of crude tricyclic *cis-*α-aminonitrile 18 to α-substituted amines could be achieved through the Bruylants reaction.? This strategy was not only used to examine the quality of the product but also provided a route to lepadiformine analogues. Thus, direct treatment of the crude aminonitrile 18 with Grignard reagents, ethynylmagnesium chloride (HCCMgCl), and trimethylsilylmethylmagnesium chloride (TMSCH_2_MgCl) afforded the α-substituted amines adducts 19 in 34% yield and 20 in 57% yield. Subsequent hydrolysis of α-amino nitrile 18 to the corresponding acid or carboxyl derivative turned out to be more challenging than expected. A number of hydrolysis conditions including acid, basic, or transition-metal-mediated or even under sealed or microwave-assisted forced conditions were examined (see Supporting Information), but none of these conditions could provide the desired products. Eventually, we found that Rychnovsky’s conditions of acidic methanolysis? were effective to form methyl ester 21 in 33% overall yield from tricyclic lactam 2. Completion of the synthesis was accomplished by LiAlH_4_ reduction of methyl ester 21 to produce alcohol 1b in 90% yield, whose spectral data were in accordance with the reported values of lepadiformine B.?
In conclusion, we developed an efficient protocol for the preparation of both cis- and trans-2-alkyl-8a-CDHQ, key intermediates for the syntheses of lepadiformine and fasicularin, through BF_3_·OEt_2_-initiated deprotectively silane-mediated alkylative/reductive cyclization. Cbz-protected cis-8a-2-allylCDHQ (4) can be achieved in 3 steps with 64% yield, while acetyl-protected trans-8a-2-n-butylCDHQ (9) can be achieved in 4 steps with 49% yield. Compared with our previous results in which 4 was obtained in 37% yield over 5 steps and 9 in 20% yield over 9 steps, this methodology facilitates the rapid and efficient assembly of two 2-alkyl-8a-CDHQ systems for the total synthesis of marine tricyclic alkaloids lepadiformine and fasicularin.
It usage was illustrated by the total syntheses of (±)-lepadiformine B and C. Lepadiformine B (1b) can be accomplished in 14 steps with 3.5% overall yield from cyclohexanone, while lepadiformine C (1c) can be accomplished in 12 steps with 9% overall yield. In addition to more efficient transformations, these reactions are readily scalable and operationally simple, not requiring harsh conditions. Various lepadiformine B analogues, such as 19 and 20, are also achieved, as demonstrated. The work provides a new approach toward the ascidian family alkaloids, and these results will be reported in due course.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sauviat M.-P.Vercauteren J.Grimaud N.Juge M.Nabil M.Petit J.-Y.Biard J.-F.Sensitivity of Cardiac Background Inward Rectifying K+ Outward Current (IK 1) to the Alkaloids Lepadiformines A, B, and CJ. Nat. Prod.20066955856210.1021/np 050215 s 16643025 · doi ↗ · pubmed ↗
- 2JugéM.Grimaud N.Biard J.-F.Sauviat M.-P.Nabil M.Verbist J.-F.Petit J.-Y.Cardiovascular effects of lepadiformine, an alkaloid isolated from the ascidians Clavelina lepadiformis (Müller) and C. moluccensis (Sluiter)Toxicon 2001391231123710.1016/S 0041-0101(01)00079-411306135 · doi ↗ · pubmed ↗
- 3Biard J. F.Guyot S.Roumkis C.Verbist J. F.Vercauteren J.Weber J. F.Boukef K.Lepadiformine, a new Marine Cytotoxic Alkaloid from Clavelina lepadiformis Muller Tetrahedron Lett.1994352691269410.1016/S 0040-4039(00)77007-9 · doi ↗
- 4Abe H.Aoyagi S.Kibayashi C.First Total Synthesis of the Marine Alkaloids (−)-Fasicularin and (−)-Lepadiformine Based on Stereocontrolled Intramolecular Acylnitroso-Diels-Alder Reaction J. Am. Chem. Soc.20001224583459210.1021/ja 9939284 · doi ↗
- 5a Greshock T. J.Funk R. L.Total Synthesis of (±)-Lepadiformine via an Amidoacrolein Cycloaddition Org. Lett.200133511351410.1021/ol 016590311678695 · doi ↗ · pubmed ↗
- 6a Abe H.Aoyagi S.Kibayashi C.Total Synthesis of the Natural Enantiomer of (−)-Lepadiformine and Determination of Its Absolute Stereochemistry Angew. Chem., Int. Ed.2002413017302010.1002/1521-3773(20020816)41:16<3017::AID-ANIE 3017>3.0.CO;2-112203443 · doi ↗ · pubmed ↗
- 7a Weinreb S. M.Lepadiformine: A Case Study of the Value of Total Synthesis in Natural Product Structure Elucidation Acc. Chem. Res.200336596510.1021/ar 020040312534305 · doi ↗ · pubmed ↗
- 8Wu J.-L.Chiou W.-H.Diastereocontrolled Formal Syntheses of (±)-Lepadiformines A, B, and C and the Divergent Synthesis of 2-epi-Lepadiformine C through Unexpected Double Consecutive Epimerizations J. Org. Chem.2020859051906310.1021/acs.joc.0c 0096432580551 · doi ↗ · pubmed ↗