X26nt-mediated recruitment of eIF4A2 facilitates CCND1 translation to drive endothelial cell cycle progression
Xinghe Zhao, Xiaocui Chen, Zheyi Chen, Lisong Shen, Lingfang Zeng, Junyao Yang

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
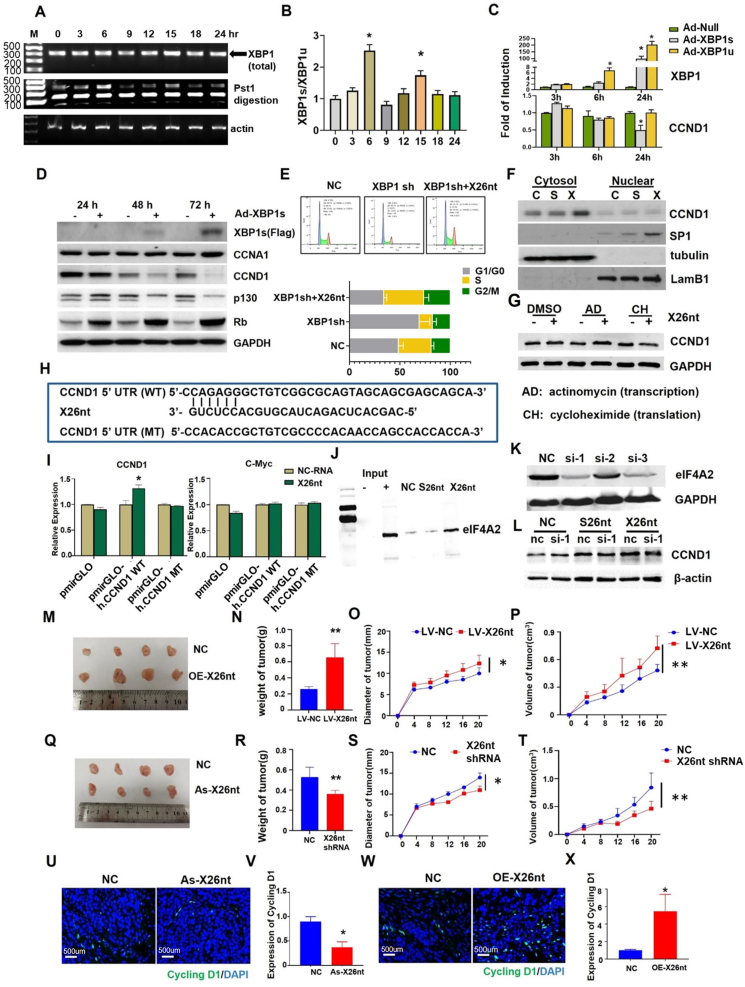 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPI3K/AKT/mTOR signaling in cancer · Mast cells and histamine · Adenosine and Purinergic Signaling
Dysregulation of cell cycle control plays a pivotal role in tumor progression. Acting as a crucial regulator of the G1/S phase transition, dysregulation of cyclin D1 (CCND1) can disrupt the delicate balance between cell proliferation and quiescence, leading to uncontrolled cell cycle progression.1 Endoplasmic reticulum stress influences the cell cycle through myriad pathways. During endoplasmic reticulum stress, the inositol-requiring enzyme 1 alpha (IRE1α) can be activated, and its endoribonuclease domain cleaves unspliced X-box binding protein 1 (XBP1u) mRNA, generating a spliced XBP1 (XBP1s) and a non-coding RNA, whose length is 26 nt (named exosomal 26-nt-long ncRNA, X26nt).2^,^3 Studies have demonstrated that XBP1s can induce the expression of pro-angiogenic factors, promoting the formation of new blood vessels.4^,^5 Our previous study demonstrated that gastric cancer exosome-derived X26nt induces tumor-associated angiogenesis via promoting endothelial cell migration targeting vascular endothelial-cadherin.2 However, the exact mechanisms by which XBP1 splicing regulates endothelial cell proliferation are still not fully understood.
When synchronizing human umbilical vein endothelial cells (HUVECs) at the late G1 phase by double thymidine block and then releasing it to re-enter the cell cycle, we observed an increase in the number of cells in the G2/M phase at 6 h and 9 h after release (Fig. S1A). We collected samples at the same indicated time points and performed PCR and Western blot analysis. The results showed that the expression level of the spliced form of XBP1 (XBP1s) was consistently increased at 6 h and 15 h (Fig. 1A, B; Fig. S1B). Vascular endothelial growth factor (VEGF) induced endoplasmic reticulum stress resulting in up-regulation of the cell cycle-related protein CCND1. Upon XBP1 knockdown or inhibition of XBP1 splicing (IRE1α knockdown), VEGF-induced up-regulation of CCND1 was eliminated (Fig. S2). Based on this, we hypothesized that XBP1 splicing could play a crucial role in mediating cell cycle in HUVECs. However, further experiments revealed that over-expression of XBP1s (Ad-XBP1s) suppressed the expression of CCND1 and other cell cycle-related proteins in HUVECs (Fig. 1C, D; Fig. S3). These findings indicated that XBP1s may not be the key factor regulating cell cycle-related protein CCND1. Thus, we focused on X26nt and hypothesized that it could act as a mediator of the cell cycle. Knockdown of XBP1 significantly reduced cell proliferation, and its X26nt, not XBP1s, could restore the proliferation capability (Fig. S4).Figure 1X26 nt can increase CCND1 through binding to the 5′ UTR of CCND1 mRNA mediated by EIF4A2, facilitate the proliferation of HUVECs, promote tumor angiogenesis, and therefore contribute to tumor progression. (A) HUVECs were synchronized at the late G1 phase by double thymidine block, and then released to re-enter the cell cycle. Cells were harvested at the time indicated after release. PCR analysis of XBP1 (total) and XBP1s expression after release at different times (n = 3). (B) Quantitative analysis XBP1s/XBP1u rate of (A). (C) HUVECs were infected with Ad-XBP1s and Ad-XBP1u, and XBP1 and CCND1 expression were analyzed by PCR. Ad-null was introduced as a control. (D) Western blots of XBP1s, CCNA1, CCND1, P130, RB, and E2F2 of Ad-XBP1s-infected-HUVECs were collected at 24 h, 48 h, and 72 h. GAPDH was introduced as the loading control. (E) Flow cytometry analysis showed the cell cycle distribution of HUVECs transfected with negative control (NC) or XBP1 shRNA, and cells transfected with XBP1 shRNA cocultured with X26nt. Bar graphs show the percentages of HUVECs in the G0/G1, S, and G2/M phases. (F) Western blot analysis of cytosol and nuclear proteins collected from HUVECs cocultured with negative control, S26nt, or X26nt. TUBULIN and LAMB1 were introduced as the loading controls. (G) The protein expression of CCND1 with the treatment of actinomycin (AD) and cycloheximide (CH) in HUVECs cocultured with or without X26nt. (H) The schematic showing potential X26nt binding sites in the CCND1 mRNA 5′ UTR. (I) The luciferase assay of HUVECs co-transfected firefly luciferase reporter plasmid containing either wild-type (pmirGLO-h. CCND1 WT) or mutant CCND1 mRNA 5′ UTR with pmirGLO-h. CCND1 mRNA cocultured with X26nt or NC-RNA. ∗p < 0.05. (J) Western blots of EIF4A2 in cells cocultured with NC, S26nt, and X26nt. (K) Immunoblots of EIF4A2 in HUVECs transfected with three different si-EIF4A2 plasmids. The scrambled EIF4A2 inhibitors were transfected as the control (nc). (L) Western blot analysis of CCND1 in nc and si-1 HUVECs co-cultured with S26nt and X26nt. β-ACTIN was introduced as the loading control. (M–P) The formed tumors from BGC-823 cells transfected with X26nt-overexpressing (OE-X26nt) lentivirus and control lentivirus (Control) were isolated and compared. Analysis of tumor diameter, volume, and weight in each group is shown. (Q–T) The BGC-823 cells were subcutaneously injected into the BALB/c nude mice to create a tumor-implanted model. When the tumor appeared, X26nt antisense RNA (As-X26nt) plasmid and mock control (NC) were injected into the tumor every other day before the mice were sacrificed. Analysis of tumor diameter volume and weight in each group is shown. (U–X) Immunofluorescence staining of CCND1/DAPI in tumors treated with X26nt asRNA plasmid and NC, as well as X26nt OE lentivirus and control. ∗p < 0.05 and ∗∗p < 0.01 versus the control. Columns, mean (B–G, I–L: n = 3; M–X, n = 4); bars, standard deviations. The data presented are representative or average of three independent experiments. X26nt, exosomal 26-nt-long ncRNA; CCND1, cyclin D1; EIF4A2, eukaryotic translation initiation factor 4A2; XBP1, X-box binding protein 1; XBP1s, spliced X-box binding protein 1; CCNA1, cyclin A1; E2F2, E2F transcription factor 2; LAMB1, laminin subunit beta 1.Figure 1
Our former studies showed that X26nt could promote the proliferation, migration, and tube formation of endothelial cells.3 To investigate the role of X26nt in the proliferation of HUVECs, we conducted loss-of-function assays by adding X26nt to XBP1s knockdown cells and observed that X26nt significantly reversed the cell cycle arrest in the G1/G0 phase induced by XBP1s knockdown cells (Fig. 1E; Fig. S5A). We also constructed a mutant S26nt as a negative control for X26nt and examined the protein levels of CCND1 and specificity protein 1 (SP1) in cytoplasmic and nuclear fractions within S26nt or X26nt overexpression (Fig. 1F). The findings confirmed that X26nt genuinely enhanced the cytoplasmic expression of CCND1. The expression of X26nt was down-regulated in Ad-XBP1s, which indicated that X26nt was the effective factor in promoting the expression of CCND1 and cell cycle (Fig. S5B). To explore the mechanism underlying the promotion of CCND1 by X26nt, we treated HUVECs with actinomycin D and cycloheximide. Actinomycin D can inhibit RNA formation and therefore reflect the efficiency of transcription. Cycloheximide can modulate translation efficiency by inhibiting protein production. The results demonstrated that X26nt could still enhance the expression of CCND1 after RNA transcription was inhibited, but its effect was lost when protein translation was inhibited (Fig. 1G; Fig. S6A). This suggests that X26nt may regulate the translation efficiency of CCND1.
We conducted further investigations to understand the mechanism underlying X26nt interacting with CCND1. Using bioinformatics tools, we predicted the binding site of X26nt on 5′ UTR of CCND1 mRNA (Fig. 1H). Subsequently, a dual-luciferase reporter assay demonstrated that X26nt significantly enhanced the activity of firefly luciferase reporter containing the wild-type 5′ UTR of CCND1 mRNA. Furthermore, this effect was abolished when the predicted binding site in 5′ UTR was mutated. Interestingly, the expression level of C-MYC was not affected by either X26nt or mutated 5′ UTR of CCND1 mRNA (Fig. 1I). To further validate the direct protein target of X26nt in proliferating endothelial cells, we synthesized biotinylated X26nt (Bio-X26nt), incubated it with VEGF-treated endothelial cells, and performed affinity purification using streptavidin beads. Mass spectrometry analysis demonstrated that eukaryotic translation initiation factor 4A2 (EIF4A2) exhibited a higher score in the VEGF-treated group, indicating its potential involvement in X26nt-mediated effects. Importantly, EIF4A2 is known to play a critical role in protein translation. This observation was further substantiated by Western blot analysis, which displayed the formation of a complex involving X26nt and EIF4A2 (Fig. 1J; Fig. S7). To substantiate that X26nt could trigger CCND1 via its interaction with EIF4A2, we utilized siRNA to suppress EIF4A2 (Fig. 1K). The findings revealed that the capacity of X26nt to enhance CCND1 expression was nullified when EIF4A2 was suppressed, corroborating our initial hypothesis (Fig. 1L; Fig. S6B).
Our former research also found that X26nt was significantly higher in gastric cancer serum and tissues, and X26nt accelerated tumor angiogenesis.3 Furthermore, we found that the levels of X26nt were remarkably higher in gastric cancer cells than in HUVECs (Fig. S8). To ascertain the role of X26nt in promoting tumor growth, a tumor-implanted model was constructed by subcutaneously injecting loss-of-function (via shRNA lentivirus-mediated knockdown of X26nt) and gain-of-function (via adenoviral transfer of X26nt) BGC-823 cells (human gastric adenocarcinoma cell line) into the back of the armpit of nude mice. Analysis of changes in tumor weight and volume trends revealed that X26nt overexpression promoted tumor growth (Fig. 1M−P), while its knockdown had the opposite effect (Fig. 1Q–T). Additionally, we noted that X26nt knockdown resulted in a decrease in CCND1, while inversely, X26nt overexpression led to an increase in CCND1 (Fig. 1U–X). Consequently, we concluded that X26nt could increase CCND1 expression and promote tumor growth in vivo.
Our former study also revealed that X26nt could inhibit the expression of vascular endothelial-cadherin and enhance the migration and tube formation abilities of endothelial cells.3 Our subsequent results confirmed that loss of X26nt induced cell cycle arrest in the G1/G0 phase. Our discovery showed that X26nt could enhance the translation efficiency of CCND1 by binding to the 5′ UTR of CCND1 mRNA, with the involvement of EIF4A2. This could promote the cell cycle progression of HUVECs in the late G1/S phase. In vivo experiments further confirmed that X26nt accelerated the expression of CCND1 and tumor growth. As far as we are concerned, there is no evidence showing that the increasing level of X26nt has a direct effect on the cell cycle. In our prior research, we did observe that the levels of X26nt were significantly elevated in the serum from gastric cancer patients compared with normal serum. Hence, we propose that the overexpressed X26nt might be secreted via gastric cancer cell-derived exosomes and subsequently internalized by HUVECs.3 This could lead to the proliferation and migration of endothelial cells, thereby encouraging tumor angiogenesis (Fig. S9). Ultimately, X26nt may be a weapon for tumor cells to modify the tumor microenvironment, and it may serve as a potential target for tumor therapy.
CRediT authorship contribution statement
Xinghe Zhao: Visualization, Investigation. Xiaocui Chen: Methodology, Investigation, Formal analysis, Data curation. Zheyi Chen: Methodology, Investigation, Formal analysis, Data curation. Lisong Shen: Writing – review & editing, Writing – original draft, Resources, Funding acquisition, Formal analysis, Conceptualization. Lingfang Zeng: Writing – review & editing, Formal analysis, Data curation, Conceptualization. Junyao Yang: Writing – review & editing, Writing – original draft, Investigation, Funding acquisition, Formal analysis, Conceptualization.
Ethics declaration
The animal study protocol was approved by the Institutional Review Board of the Laboratory Animal Ethical and Welfare Committee Xinhua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine (approval number: XHEC-F-2024-002, date of approval: 2024-01-30).
Funding
This research was funded by the 10.13039/501100001809National Natural Science Foundation of China (No. 81802082 to Junyao Yang; 81672363 to Lisong Shen), the Natural Science Foundation of Shanghai Science and Technology Innovation Action Plan (China) (No. 21ZR1441500 to Junyao Yang), and the Shanghai "Rising Stars of Medical Talent" Youth Development Program-Clinical Laboratory Practitioners Program (China) (No. 2019016 to Junyao Yang).
Conflict of interests
The authors declared no conflict of interests. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bustany S.Cahu J.Guardiola P.Sola B.Cyclin D 1 sensitizes myeloma cells to endoplasmic reticulum stress-mediated apoptosis by activating the unfolded protein response pathway BMC Cancer 1520152622588129910.1186/s 12885-015-1240-y PMC 4399746 · doi ↗ · pubmed ↗
- 2Yang J.Xu J.Danniel M.The interaction between XBP 1 and ENOS contributes to endothelial cell migration Exp Cell Res 363220182622702935298710.1016/j.yexcr.2018.01.016 · doi ↗ · pubmed ↗
- 3Chen X.Zhang S.Du K.Gastric cancer-secreted exosomal X 26nt increases angiogenesis and vascular permeability by targeting VE-cadherin Cancer Sci 11252021183918523320556710.1111/cas.14740 PMC 8088954 · doi ↗ · pubmed ↗
- 4Martin D.Li Y.Yang J.Unspliced X-box-binding protein 1 (XBP 1) protects endothelial cells from oxidative stress through interaction with histone deacetylase 3J Biol Chem 28944201430625306342519080310.1074/jbc.M 114.571984 PMC 4215241 · doi ↗ · pubmed ↗
- 5Zeng L.Xiao Q.Chen M.Vascular endothelial cell growth-activated XBP 1 splicing in endothelial cells is crucial for angiogenesis Circulation 127162013171217222352961010.1161/CIRCULATIONAHA.112.001337 · doi ↗ · pubmed ↗