The diagnostic accuracy of ultrasound and genomic tests for the diagnosis of autosomal-dominant polycystic kidney disease: a systematic mapping review
Sue Harnan, Matthew Gittus, Louise Falzon, Miranda Durkie, Olena Mandrik, Albert C Ong, James Fotheringham

TL;DR
This review compares the accuracy of ultrasound and genomic tests for diagnosing autosomal-dominant polycystic kidney disease and finds that test performance varies with age and genetic factors.
Contribution
The study provides a systematic mapping of test accuracy trends and characteristics for ADPKD diagnosis over time.
Findings
Genomic test sensitivity remained around 78% despite technological advances from 2000 to 2023.
Ultrasound accuracy improves with age and is lower in PKD2 patients compared to PKD1.
No genomic studies were found in first-degree relatives of ADPKD patients.
Abstract
Genomic and ultrasound tests can provide diagnostic and prognostic information on autosomal-dominant polycystic kidney disease (ADPKD), and can screen first-degree relatives in whom early diagnosis can be advantageous. We conducted a systematic mapping review on test accuracy and characteristics over time. Medline, Embase, and Cochrane were searched (August 2023) for studies in first-degree relatives/individuals clinically diagnosed with ADPKD receiving genomic or ultrasound tests. Acceptable reference standards for sensitivity/detection rate and specificity were definitive imaging or genomic confirmation. Genomic studies were categorized by technology and read length. Relationships between sensitivity, specificity, genomic technology, diagnostic criteria/reference standard, and genes tested were compared. From 1029 non-duplicate titles retrieved, 51 genomic and 7 ultrasound studies…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
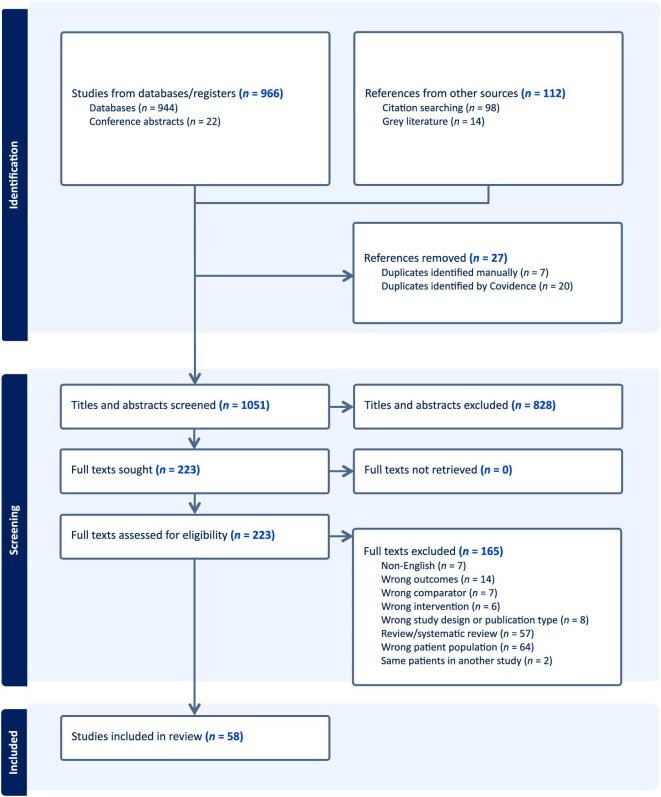 Figure 1
Figure 1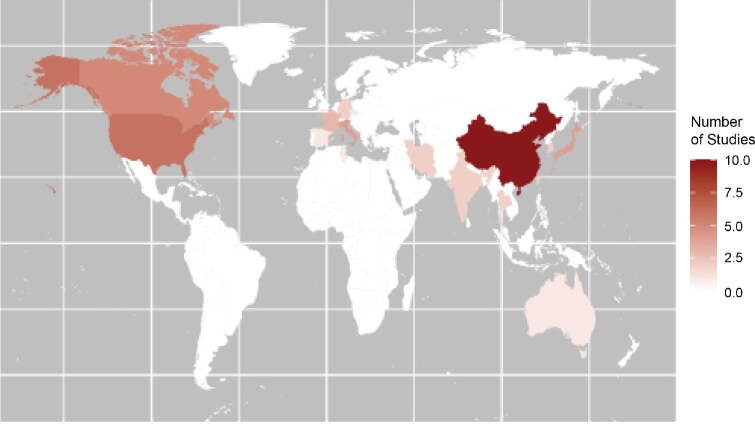 Figure 2
Figure 2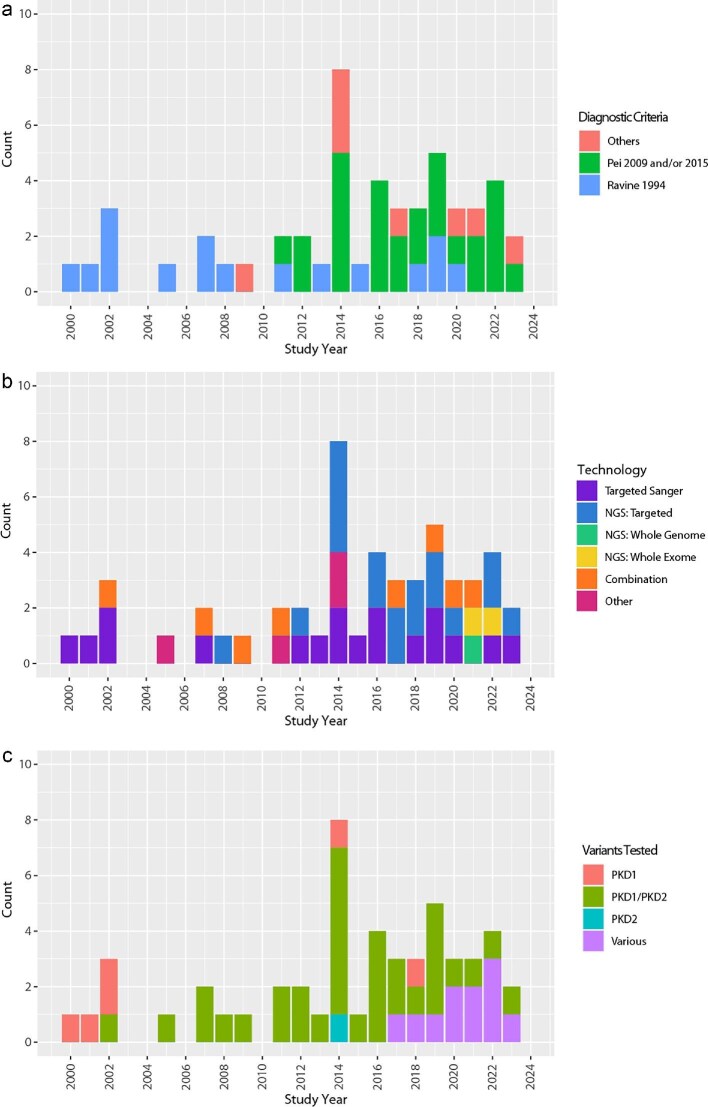 Figure 3
Figure 3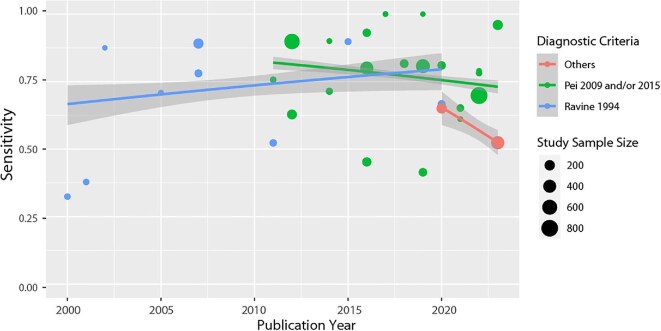 Figure 4
Figure 4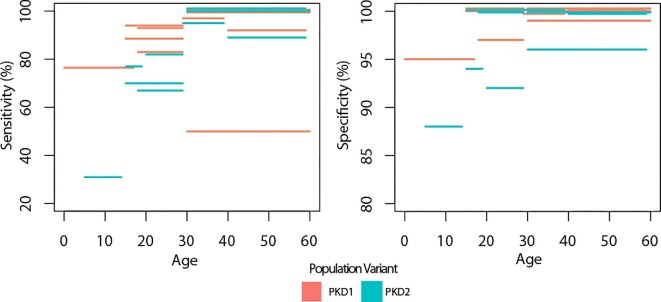 Figure 5
Figure 5| Genomic test or strategy utilizing genomic test(s) | Grey-scale ultrasound | ||
|---|---|---|---|
| Population | 1st degree relatives of patients with an ADPKD diagnosis (clinical or genomic) | People with a clinical ADPKD diagnosis according to Pei | 1st degree relatives of patients with an ADPKD diagnosis (clinical or genomic) |
| Index test | Genomic test or diagnostic strategy utilizing genomic tests | Grey-scale ultrasound | |
| Reference standard | Imaging according to Pei | Imaging according to Pei | |
| Target condition | ADPKD | ||
| Outcome | ● Sensitivity and specificity; TP, FP, TN, FN. If not available, diagnostic rate (sensitivity) | ||
| Study design | Diagnostic test accuracy studies. If none available, studies reporting sensitivity only were eligible. | ||
| Components of genomic testing | Definition |
|---|---|
| Technology | The specific tools and platforms used to sequence and analyse DNA |
| Read length | The length of DNA sequence read by a sequencing machine in a single run, typically ranging from 50 to several thousand base pairs |
| Enrichment method | Techniques used to selectively capture and sequence specific regions of the genome |
| Analysis | Computational processes and algorithms used to interpret raw sequencing data including the examination of specific sets of genes |
| Genomic structural variation analysis | The identification of changes such as deletions, insertions, inversions, translocations, single nucleotide variations, and copy number variations |
| Author, year, country |
| Proband/per family? | Family history | Reference standard | Genes targeted | Enrichment method | Small sequence variant analysis | Copy number variant analysis |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Pei 2009 recruitment criteria ( | ||||||||
| Ali 2019 [ | 6 | Proband or per family | With family history | Pei 2009 |
| Amplicon | NA | None |
| Audrezet 2012 [ | 528 (with FH) | Proband or per family | With family history | Pei 2009 |
| Amplicon | NA | None |
| Hwang 2016 [ | 220 | Proband or per family | Either with or without family history | Pei 2009 |
| Amplicon | NA | MLPA |
| Li 2022 [ | 19 | Proband or per family | Either with or without family history | Pei 2009 |
| Amplicon | NA | MLPA |
| Pei 2009 and Torres 2012 recruitment criteria ( | ||||||||
| Liu 2014a [ | 10 | Proband or per family | Either with or without family history | Family history: Pei 2009; no family history, Torres 2012 |
| Amplicon | NA | MLPA |
| Pei 2009/2015 recruitment criteria ( | ||||||||
| Carrera 2016 [ | 440 | Proband or per family | Either with or without family history | Pei 2009/2015 |
| Amplicon | NA | MLPA |
| Orisio 2023 [ | 198 | Proband or per family | Either with or without family history | Pei 2009/2015 |
| Amplicon | NA | MLPA |
| Ravine 1994 recruitment criteria ( | ||||||||
| Abdelwahed 2018 [ | 18 | Some related | Either with or without family history | Ravine 1994 |
| Amplicon | NA | MLPA |
| Burtey 2002 [ | Proband or per family | Either with or without family history | Criteria equivalent to Ravine 1994 |
| Amplicon | NA | None | |
| Chang 2013 [ | 46 | Proband or per family | Either with or without family history | Ravine 1994 |
| Amplicon | NA | MLPA |
| Garcia-Gonzalez 2007 [ | 82 | Proband or per family | Either with or without family history | Ravine 1994 |
| Amplicon | NA | None |
| Inoue 2002 [ | 8 | Proband or per family | Not reported | Ravine 1994 |
| Amplicon | NA | None |
| Liu 2015 [ | 49 | Proband or per family | Either with or without family history | Ravine 1994 |
| Amplicon | NA | MLPA |
| Pandita 2019 [ | 125 | Proband or per family | Either with or without family history | Ravine 1994 |
| Amplicon | NA | MLPA |
| Phakdeekitcharoen 2000 [ | 37 | Proband or per family | Not reported | Ravine 1994 |
| Amplicon | NA | None |
| Phakdeekitcharoen 2001 [ | 37 | Proband or per family | Not reported | Ravine 1994 |
| Amplicon | NA | None |
| Raj 2020 [ | 84 | Proband or per family | Either with or without family history | Ravine 1994 |
| Amplicon | NA | None |
| Genomic recruitment criteria ( | ||||||||
| Liu 2014b [ | 8 | Proband or per family | Not reported | Sanger genotyping | PKD1; PKD2 | Amplicon | NA | None |
|
| ||||||||
| Pei 2009 recruitment criteria ( | ||||||||
| Choi 2014 [ | 20 | Proband or per family | Either with or without family history | Pei 2009 |
| Hybridization | Targeted panel | MLPA |
| Jin 2016 [ | 148 | Unclear | Either with or without family history | Pei 2009 |
| Hybridization | Targeted panel | None |
| Kinoshita 2016 [ | 101 | Proband or per family | Not reported | Pei 2009 |
| Amplicon | Targeted panel | MLPA |
| Mochizuki 2019 [ | 111 | Unclear | Not reported | Pei 2009 |
| Hybridization + Amplicon | Targeted panel | MLPA |
| Ranjzad 2017 [ | 18 | Proband or per family | With family history | Pei 2009 |
| Hybridization | Targeted panel | None |
| Rossetti 2012 [ | 183 | Proband or per family | Not reported | Pei 2009 |
| Amplicon | Targeted panel | None |
| Tan 2014 [ | 25; 3; 25 | Unclear | Not reported | Sanger sequencing |
| Amplicon | Targeted panel | None |
| Xu 2018 [ | 120 | Proband or per family | Not reported | Pei 2009 |
| Amplicon | Targeted panel | MLPA |
| Yang 2014 [ | 7 | Proband or per family | Either with or without family history | Pei 2009 |
| Amplicon | Targeted panel | None |
| Yu 2022 [ | 882 | Proband or per family | Either with or without family history | Pei 2009 |
| Amplicon | Targeted panel | None |
| Pei 2015 recruitment criteria ( | ||||||||
| Hosseinpour 2022 [ | 32 | Proband or per family | Either with or without family history | Pei 2015 |
| Hybridization | Targeted panel | None |
| Ravine 1994 recruitment criteria ( | ||||||||
| Zhang 2019 [ | 62 | Proband or per family | Not reported | Ravine 1994 |
| Hybridization | Targeted panel | MLPA |
| Zhao 2008 [ | 3 | Proband or per family | Either with or without family history | Ravine 1994 in Probands |
| Amplicon | Targeted panel | None |
| Pei 2009/Torres recruitment criteria ( | ||||||||
| Fujimaru 2018 [ | 53 | Proband or per family | Without family history | CT or MRI (>10 cysts in each kidney), Pei 2009, Torres 2012, Torres 2017 | 69 genes causing hereditary renal cystic disease | Hybridization | Targeted panel | NGS CNV |
| Other imaging recruitment criteria | ||||||||
| Lindemann 2023 [ | 441 | Unclear | Either with or without family history | Imaging as per footnote |
| Amplicon | Targeted panel | None |
| Mantovani 2020 [ | 191 | Unclear | Either with or without family history | Pei 2009 criteria for US, MRI, CT if equivocal |
| Amplicon | Targeted panel | MLPA |
| Genomic criteria ( | ||||||||
| Trujillano 2014 [ | 36 | Unclear | Not reported | Sanger sequencing of |
| Amplicon | Targeted panel | None |
|
| ||||||||
| Genomic recruitment criteria ( | ||||||||
| Borras 2017 [ | 19 | Proband/per family | NR | Genomic (short read WGS or WES) |
| SMRT | Panel | NGS breakpoint detection, MLPA |
|
| ||||||||
| Pei 2009 recruitment criteria ( | ||||||||
| Mallawaarachchi 2021 [ | 42 | Proband/per family | Either with or without family history | Pei 2009 |
| Hybridization | Virtual panel | NGS CNV, MLPA |
|
| ||||||||
| Pei 2009 and wider atypical disease recruitment criteria ( | ||||||||
| Chang 2022 [ | 235 | Unclear | Either with or without family history | Pei 2009 |
| Hybridization | Virtual panel | NGS CNV |
| Elliott 2021 [ | 18 | Proband or per family | Either with or without family history | Typical ADPKD: Pei 2009 | Typical ADPKD: | Hybridization | Virtual panel | None |
|
| ||||||||
| DHPLC then first generation: targeted Sanger sequencing ( | ||||||||
| Ravine 1994 recruitment criteria ( | ||||||||
| Rossetti 2002 [ | 45 | Proband or per family | Not reported | Ravine 1994 criteria |
| Amplicon | NA | None |
| Rossetti 2007 [ | 127 | Proband or per family | Not reported | Ravine 1994 |
| Amplicon | NA | None |
| Yu 2011 [ | 65 | Proband or per family | Either with or without family history | Ravine 1994 |
| Amplicon | NA | None |
| Genomic recruitment criteria ( | ||||||||
| Tan 2009 [ | 14 | Unclear | Not reported | Unclear ‘PKD genotyping’ by reference lab |
| Hybridization | NA | None |
|
| ||||||||
| KDIGO guidelines (Chapman 2015) ( | ||||||||
| Hu 2021 [ | 26 | Proband or per family | Either with or without family history | KDIGO guidelines (Chapman 2015) | Tier 1: WES and | Hybridization | Targeted Panel/NA | MLPA |
|
| ||||||||
| Pei 2009/2015 recruitment criteria ( | ||||||||
| Schonauer 2020 [ | 100 | Some related | Either with or without family history | Pei 2009 |
| Hybridization | Targeted Panel/Virtual Panel | MLPA |
|
| ||||||||
| Pei 2009/2015 recruitment criteria ( | ||||||||
| Iliuta 2017 [ | 205 | Proband or per family | Either with or without family history (and reported separately) | Pei 2009 |
| Hybridization | NA/Targeted Panel | None |
|
| ||||||||
| Pei 2009 recruitment criteria ( | ||||||||
| Kim 2019 [ | 524 | Proband or per family | Either with or without family history | Pei 2009 |
| Hybridization | Targeted Panel/NA | MLPA |
|
| ||||||||
| HRM ( | ||||||||
| Pei 2009 recruitment criteria ( | ||||||||
| Bataille 2011 [ | 37 | Proband or per family | Not reported | Pei 2009 |
| NA | NA | None |
| Obeidova 2014 [ | 56 | Proband or per family | Either with or without family history | Pei 2009 |
| NA | NA | MLPA |
| PKD-2 linkage analysis recruitment criteria ( | ||||||||
| Virzi 2014 [ | 16 | Proband or per family | Not reported |
|
| NA | NA | None |
|
| ||||||||
| Ravine 1994 recruitment criteria ( | ||||||||
| Zhang 2005 [ | 24 | Proband or per family | Not reported | Ravine 1994 |
| NA | NA | None |
| Author, year, country | Population |
| Index test criteria for ADPKD | Ultrasound technology | Reference standard* | Age bands reported | Gene subgroups reported |
|---|---|---|---|---|---|---|---|
|
| |||||||
| First-degree relatives | |||||||
| Parfrey 1990 [ | 1st degree family members of | 126 people from 10 PKD1 families | 1+/2+ | NR | Gene linkage analysis | </> 30 years | All were |
| Elles 1994 [ | 1st degree relatives of ADPKD (criteria for probands unclear) | 80 | Bear 1984 | 3.5-MHz scanner | Genomic markers | </> 30 years | Only reports results for |
| Ravine 1994 [ | Undiagnosed 1st degree relatives of confirmed | 204 (from 18 families) | 1+/2+ | 3- or 5-MHz | >95% or <5% probability of PKD1 by DNA linkage analysis | 15–29 | All were |
| Gabow 1997 [ | Children 1st degree relatives of ADPKD1 families (genomically confirmed) | 106 children (from 40 families) | Any cysts | NR | Gene linkage analysis | Children | All were |
| Nicolau 1999 [ | 1st degree relatives of Type 1 or Type 2 ADPKD (genomically confirmed) | 319 individuals from 54 families | Ravine 1994: | 3.7 or 5-MHz | Genetic linkage study | </>30 years |
|
| Demetriou 2000 [ | 1st degree relatives of ADPKD Type 2 families | 211 alive people at risk from 3 families | Ravine 1994 (ADPKD-1) for adults and Gabow 1997 for children 5–14: 1+ Cysts 15–19: 1+/1+ Cysts Or 2+/020–29: 2+/1+ (3+ and bilateral involvement) | 3.5 or 5-MHz | DNA linkage and direct mutation analyses | 5–14 | All were |
| Pei 2009 [ | 1st degree relatives at risk of PKD1 or PKD2 (proband diagnostic criteria unclear) | 948 | 15–39: 3+ total | 3- or 5-MHz | Genomic testing (range of methods) | 15–29 |
|
- —National Institute for Health and Care Research10.13039/501100000272
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and Kidney Cyst Diseases · Renal and related cancers · Genetic Syndromes and Imprinting
INTRODUCTION
Autosomal-dominant polycystic kidney disease (ADPKD) is the most common hereditary kidney disease, affecting an estimated 12 million individuals worldwide [1]. Being dominantly inherited, first-degree relatives have a 50% risk of developing the condition [2]. It is characterized by cystic expansion of the kidneys, progressing to bilateral kidney enlargement and subsequent chronic kidney disease (CKD) [3]. Symptoms typically begin around age 30 [4], and 50% of people with ADPKD require kidney replacement therapy by age 60 [5]. Although ADPKD is primarily caused by variants in PKD1 and PKD2 genes, ongoing discoveries of other causative genes have revealed greater genomic heterogeneity than previously understood [3, 6]. Even within PKD1 and PKD2 genes, there is significant allelic heterogeneity with >1200 and almost 190 pathogenic/likely pathogenic variants identified for PKD1 and PKD2, respectively [7]. Most identified families have unique variants, with fewer than 2% of unrelated ADPKD-affected families sharing the same variant [8].
ADPKD diagnosis is mostly based on imaging and family history, and it can be difficult to differentiate from other cystic kidney diseases when imaging results are atypical or in young individuals with a negative family history [6]. By age 40, a diagnosis of ADPKD can be ruled out in people who have no more than one kidney cyst [9]. Genomic testing can provide a definitive diagnosis for patients, relatives at risk of inheriting the disease, and for individuals who are seeking genomic consultation prior to pre-implantation genomic diagnosis for reproduction or living kidney donor transplantation [6]. If possible, genomic testing of a family member who has a clinical diagnosis of ADPKD using a full diagnostic genomic test, usually including PKD1 and PKD2 genes as a minimum, is the recommended first step when genomic testing individuals at risk of inheriting ADPKD is being performed. If a pathogenic variant is identified in this family member, then predictive testing in their relatives can be offered by targeted analysis of the familial pathogenic variant.
Historically, guidelines have hesitated to recommend genomic screening due to costs and limited accessibility [10]. The Kidney Disease Improving Global Outcomes (KDIGO) clinical practice guidelines state that an ultrasound diagnosis can be used even when the family is genetically resolved [11]. These guidelines have been designed to be applicable to healthcare systems worldwide and as costs associated with genomic tests drop, gene panels broaden, and technology advances, a review of contemporary evidence to inform clinical practice guidelines is required. Earlier diagnosis has the potential to enable earlier management and improve outcomes for people with ADPKD. This can occur through earlier access to lifestyle and medication interventions, family planning, and living donation information [12, 13]. This systematic mapping review aims to describe and characterize the available diagnostic accuracy literature relating to ultrasound and genomic tests for people at risk of ADPKD. We aim to look at the changes in technology and chart the sensitivity of genomic tests over time and the diagnostic accuracy of ultrasound tests, to provide an overview of this fast-paced and complex topic.
MATERIALS AND METHODS
This systematic mapping review is reported in line with recommendations made by PRISMA for scoping reviews [14], since there is no guidance for mapping reviews. We also considered relevant items from the PRISMA guidance for reporting diagnostic test accuracy reviews [15]. There is no standard definition of a mapping review [16], but they are generally descriptive in nature, do not include statistical synthesis, but rather use graphical, tabular, and narrative methodologies to characterize the literature.
The protocol was registered on the PROSPERO database (record number CRD42023456727), but some changes were made to the protocol as detailed in Online Supplement 1
Search strategy
Potentially relevant articles were identified by searching Ovid Medline, Ovid Embase, and the Cochrane Library from inception to August 2023. Relevant subject headings and free-text terms to represent ‘Autosomal Dominant Polycystic Kidney Disease’ AND ‘ultrasound’ OR ‘genetic screening’ were used. A validated search filter to identify diagnostic studies was applied [17], but the studies were not limited by year or language. Reference lists of relevant studies and reviews, and relevant articles in the Similar Articles feature in PubMed, and the Cited Reference Search in ISI Web of Science were also screened. The following relevant conferences were searched for the past 3 years: American Society of Nephrology Kidney Week, World Congress of Nephrology, and European Renal Association Congress. Full details of the search dates and strategies are available in Online Supplement 2
Study selection
The selection criteria for the review are reported in Table 1. Studies of ultrasound were included if they recruited first-degree relatives of people with ADPKD (i.e. people with 50% risk of having ADPKD) and the reference standard was imaging after age 40 years according to published criteria (e.g. Pei et al. [18], Pei et al. [19], Torres et al. [20]), or genomic confirmation by any genomic method (e.g. gene linkage analysis, Sanger sequencing). Studies using high-resolution ultrasound were excluded because standard ultrasound remains the predominant method in clinical use.
Studies of genomic tests or diagnostic strategies including genomic tests were included if they recruited either first-degree relatives of people with an ADPKD diagnosis, or people with or without a family history with a clinical ADPKD diagnosis according to published diagnostic criteria (e.g. Pei et al. [18], Pei et al. [19], Torres et al. [20]), because these are the groups the tests would be used in. The reference standard could be a diagnosis using published criteria, or a genomic diagnosis. This was a change from the published protocol because no studies met the original criterion (see Online Supplement 1).
In both reviews, prenatal populations were excluded since short follow-up meant it was not clear if all the foetuses grew up to have the disease, and the pathogenic variant may have resulted in prenatal death such that testing in a child would never have been necessary.
We did not restrict inclusion to studies using the American College of Medical Genetics and Genomics guidance for the interpretation of sequence variants [21], but attempted to standardize definitions where possible (see the section on ‘Sensitivity’).
Two reviewers (S.H. and M.G.) separately used Covidence with AI-assisted study prioritization to screen studies according to the inclusion criteria, considering first the title and abstract, then examining the full texts of the remaining articles. Any disagreements were resolved through discussion and involvement of a third reviewer (J.F.).
Data extraction and quality assessment
A data extraction form was created in Google Sheets, piloted on two articles and improved where necessary. Data extraction fields and methods are provided in Online Supplement 2 but briefly comprised data extraction, data coding, and data double-checking by a second reviewer with resolution of disagreements through discussion.
As none of the studies of genomic tests were true diagnostic test accuracy studies and were therefore of generally low quality, QUADAS 2 [22] quality assessment was not performed.
Mapping analysis
The evidence map was primarily analysed according to two main criteria:
(i)Test type: ultrasound studies were grouped separately from genomic studies. Genomic studies were then categorized according to the sequencing technology used. These sequencing technology components are defined in Table 2 and categories described in Online Supplement 3 Studies were grouped by technology used (Sanger or next generation), the genomic target (targeted gene, whole exome or whole genome), and whether the read length was short (first and second generation) or long (third generation).(ii)Population: the criteria used to recruit patients may affect the detection rate since early clinical definitions were largely based on PKD1 (Ravine [28]) and then expanded to PKD2 (Pei [18] and Pei [19]). Studies were therefore grouped according to the criteria used to define the clinical diagnosis of ADPKD. Clinical criteria included Ravine [28], Pei [18] for ultrasound, and Pei [19] for MRI or sometimes CT. Other criteria could be used for atypical presentations, such as Torres et al. [20]. Studies could cite published criteria, or accurately describe the criteria that were then matched to the corresponding citation. Studies that recruited patients according to a genomic diagnosis were grouped separately.
Several plots were then generated using R version 4.4 to show trends over time for factors including recruitment criteria, test types, gene targets, and detection rate. Changes in longitudinal detection rate were estimated using the ggplot2 generalized linear model smoothed conditional mean function, weighted for study size, with a binomial link function.
RESULTS
The search strategy retrieved a total of 1078 titles, from which 27 duplicates were removed. Of the 1051 records remaining, 828 were excluded on the basis of their title or abstract. The full text of 223 studies were assessed for eligibility, and of these 165 were excluded (see Fig. 1 for reasons). Seven studies [18, 23–28] of ultrasound and 50 studies [6, 8, 29–76] of genomic tests were included in the review.
PRISMA flow chart showing the process of study selection for the review.
Studies of genomic tests
Location of studies
The countries of origin of the included studies are mapped in Fig. 2. The country contributing the most studies was China (n = 10) [42, 46, 49, 51, 52, 71–75], followed by the USA (n = 6) [35, 63, 64, 67, 68, 77]. The remainder were from across the globe, including Canadian, European Middle Eastern, and Asian studies.
Map of origin of included studies.
Recruitment criteria
Among the 51 genomic test studies (Table 3) patients were recruited according to Pei et al. 2009 [18] and its extension Pei et al. 2015 [19] (n = 25 studies) [6, 8, 30, 31, 34, 35, 37, 38, 41, 43, 44, 46–49, 53, 55–57, 62, 65–67, 71, 72], most often. These criteria were derived in PKD1 and PKD2 patients. Ravine et al.’s [28] criteria, which targeted PKD1 patients, were used in 16 studies [29, 33, 36, 40, 45, 51, 58–61, 63, 64, 73–76]. Other imaging criteria (Torres et al. [20], Torres et al. 2017 [78], KDIGO guideline criteria) were used in a further five studies [39, 42, 50, 52, 54], and these are likely to recruit a wider population than just PKD1 and PKD2. Four studies recruited people using genomic tests: one [69] targeted people with PKD1 and PKD2 pathogenic variants and aimed to include as many different variants as possible, while the other three [32, 68, 77] did not state which genes were targeted. One study used PKD2 families previously analysed by linkage analysis [70]. Surprisingly, Ravine et al.’s 1994 [28] criteria were used to recruit patients in four studies [29, 58, 61, 74] published between 2018 to 2020. However, overall, due to the criteria used, the populations included in more recent studies were more heterogeneous and less phenotypically characteristic of PKD1/PKD2 pathogenic variants (Fig. 3a).
Charts of study characteristics over time. (a) Diagnostic criteria/radiological reference standard for inclusion of individuals with clinical diagnosis of ADPKD by year of study publication; (b) genomic test technology by year of study publication; and (c) genes analysed by genomic tests by year of study publication.
Reference standard
In nearly all cases, the reference standard was the same as the recruitment criteria. As already noted, these studies are only able to estimate detection rate (sensitivity) and cannot estimate specificity.
Test types
Among the 51 genomic test studies [6, 8, 29–77] (Table 3), there was a similar number of studies of targeted Sanger sequencing (n = 18) [8, 29, 30, 33, 34, 36, 40, 43, 45, 49, 51, 52, 57–61, 77] and targeted short read next-generation sequencing (n = 17) [6, 37, 39, 41, 46, 48, 50, 54, 55, 62, 65, 67, 69, 71, 72, 74, 76]. There was only one study of targeted long read next-generation sequencing [32], one of WGS short read next-generation sequencing [53], two of WES short read next-generation sequencing [35, 38], eight tests used a combination of technologies [42, 44, 47, 63, 64, 66, 68, 73], and four reported on other types of genomic tests [31, 56, 70, 75]. Studies were published from 2000 to 2023 (date of searches).
Figure 3b charts the types of test used over time. Sanger sequencing has been used consistently throughout the period, while the application of next-generation technologies to ADPKD diagnosis was first reported in 2008 and use has increased over time. The one study of long read technology was published in 2017 [32]. Studies on tests used in combination started in 2002, with early studies focusing on DHPLC followed by Sanger sequencing [36, 63, 64, 68, 73], and later studies mostly using combinations of next-generation sequencing, MLPA and Sanger but not always in the same order [42, 44, 47, 66]. Other test types encountered included high-resolution melt (HRM) [31, 56, 70] and single-strand conformation polymorphism analysis (SSCP) [75].
Gene targets
The genes targeted by genomic tests also broadened over time (Fig. 3c). Four of the seven studies [29, 33, 42, 45, 52, 59, 60] that only focused on PKD1 were among the five earliest studies conducted (2000 to 2002) [33, 45, 59, 60, 63]. Testing for genes beyond PKD1 started with the inclusion of PKD2 by Rossetti et al. [63], and expanded beyond PKD1 and PKD2 in 2017, when Iliuta et al. [44] included GANAB and HNF1B. Later tests [6, 38] broadened into COL4A1, DNAJB11, REN, and UMOD.
Sensitivity
Heterogeneity in the terminology used to categorize pathogenic variants supported grouping terminology erring towards the variant being pathogenic together. e.g. pathogenic, probably/likely/definitely/strong pathogenic, disease-causative, possibly damaging. To plot detection rate over time a subgroup of studies that reported both pathogenic/definitely pathogenic and likely/probably pathogenic (or similar terms) variants were selected. Studies were further grouped into three categories, to match the recruitment criteria to the genes tested (Ravine [28] criteria, only genomic tests for PKD1 or more were included; Pei 2009/2015 [19], genomic tests for PKD1 and PKD2 or more were included; other criteria, only genomic tests for PKD1, PKD2, and at least one other gene were included). Figure 4 plots the sensitivity of the tests for these three subgroups. Across all three groups, the median detection rate was 78% (interquartile range 65% to 88%, total range 32% to 100%). Sensitivity remained fairly stable over the years (Ravine [28] subgroup, range 32% [59] to 90% [51] and Pei 2009/2015 subgroup, range 41% [55] to 100%) [30, 62] or had too few points for inference (Others subgroup).
Diagnostic test accuracy (proportion with genomic variants classed as definitely pathogenic, pathogenic, likely pathogenic, and probability pathogenic or similar terms), stratified by genes targeted and recruitment criteria, by study publication year. Blue line, studies recruiting according to Ravine [28], with genomic testing for PKD1 or more; green line, studies recruiting according to Pei 2009/2015 [18, 19], with genomic testing for PKD1 and PKD2 or more; red line, studies recruiting according to other criteria, with genomic testing for more than PKD1 and PKD2. Studies weighted by size when estimating longitudinal changes and 95% confidence intervals (grey).
Ultrasound studies
The characteristics of the seven studies [18, 23–28] are outlined in Table 4. The date of studies ranged from 1990 [27] to 2009 [18] (NB Pei et al. 2015 [19] did not meet the inclusion criteria as it used high-resolution ultrasound). All [18, 23–28] recruited people at 50% risk of ADPKD from families with. PKD1 (n = 4) [24, 25, 27, 28], PKD2 (n = 1) [23], or PKD1 and PKD2 (n = 2) [18, 26] genotypes. All used a genomic reference standard.
Both sensitivity and specificity improved as age increased (see Fig. 5 and Online Supplement 4), across both PKD1 and PKD2 populations, but accuracy was poorer in PKD2 compared to PKD1 populations. The lowest sensitivity and specificity were 31% and 88%, respectively, reported in PKD2 populations aged 5–14. The highest were 100% and 100%, respectively, in multiple gene/age categories.
Sensitivity and specificity of ultrasound studies. Each bar represents the sensitivity or specificity for the age range spanned by the bar, as reported by individual studies included in this review.
DISCUSSION
Using 58 studies of genomic (n = 51) and ultrasound (n = 7) testing spanning over 30 years, this review charts the evolving methods available for screening first-degree relatives of people affected by ADPKD. Notably, none of the genomic studies we found recruited relatives at 50% risk of ADPKD, meaning the accuracy of the tests in this population is unclear. The available evidence suggests that, among people who have a clinical diagnosis of ADPKD but they or their family have not previously had genomic testing, the sensitivity of genomic tests is likely to be somewhere between 70% and 80%, depending on test methodology and the proportion of unknown variants within the population sample. Sensitivities lower than 50% and higher than 90% have also been reported.
Due to technological advances and increased sharing of known pathogenic variants we expected to see an increase in sensitivity over time, but instead the evidence suggests that the detection rate has not changed greatly. Possible explanations for these findings include (i) the small impact that increased testing of a panel of cytogenic genes has when the vast majority of pathogenic variants are in PKD1 and PKD2; (ii) pathogenic variants not detected by the methodology used e.g. deep intronic variants/structural variants/regulatory variants; (iii) unique variants detected with insufficient evidence to reach a (likely) pathogenic score (i.e. variant of uncertain significance); (iv) other cystogenic genes being responsible; (v) other causes e.g. simple age-related cysts; and (vi) the observed widening recruitment criteria leading to more atypical cases being recruited, and an increase in the size of the population for which genomic testing has become relevant, owing to the identification of additional rare ADPKD genes such as GANAB and HNF1B, COL4A1, DNAJB11, REN, and UMOD (see Table 3). However, only one study in this review included the recently identified IFT140 gene that has been shown to be the third most common associated with ADPKD after PKD1 and PKD2 [79]. The extent and rate at which current gene panels have adopted these more recently identified variants was not the subject of our study, but it is possible laboratories may not wait for extensive publication on variants before incorporating them in their gene panels.
Meanwhile, no studies on the performance of ultrasound screening in first-degree relatives of families with the more contemporary known pathogenic variants were identified. Consequently, the test accuracy of ultrasound outside populations with PKD1 and PKD2 is currently unknown. The KDIGO guidelines [11] recommend that when making an initial diagnosis of ADPKD in an adult at risk, abdominal imaging by ultrasound can be used even when the family is genetically resolved. This is despite the lack of evidence in populations outside PKD1 and PKD2. Whereas genetically unresolved families are reliant on this screening modality and guidelines continue to recommend the use of ultrasound in families with other variants, further studies are required to establish the accuracy of the test in these populations. Clinicians may need to keep these uncertainties in mind when planning further monitoring and when considering alternative diagnoses.
In clinical practice, relatives of individuals who have no pathogenic variant identified by genomic testing may be receiving radiological screening tests derived and validated in populations who broadly speaking have different pathogenic variants, since our review found all ultrasound studies recruited patient with known PKD1 or PKD2. This may lead to uncertainty in clinical diagnoses, or incorrect exclusion of disease in relatives who are still in the early stages of a clinical disease with a more slowly progressing natural history.
This systematic mapping review has been conducted to the same standards as a systematic review in terms of the search methodology, study selection, and data extraction. Data were organized according to several factors that may affect test metrics, including the recruitment criteria and reference standards used. Nevertheless, it does have some methodological limitations, often generated by the available evidence. The lack of data on diagnostic test accuracy of genomic tests in people at risk of ADPKD lead to protocol amendments including widening criteria to include studies reporting only sensitivity and in people with clinically confirmed ADPKD (removing the requirement for this to be confirmed after age 40). As a result, the included studies were not true diagnostic test accuracy studies. Critical appraisal using QUADAS-2 [22] was not performed because it is not designed for these studies and would have been uninformative. Heterogeneity in populations and test methodologies precluded meta-analysis. Since the genomic studies did not specify that included patients had to have a radiological diagnosis after a certain age, and since cysts tend to increase over time, the populations recruited according to these criteria may include more patients who presented at a young age and therefore have more progressive disease. Finally, there will inevitably remain some heterogeneity in how the pathogenic categories were defined, especially as new variants were identified and guidelines to determine variant pathogenicity have changed over time [80].
Policy makers should consider the generalisability of the patient populations recruited to the studies, which are broadening over time, to their own populations. The specifics of the test methodologies with respect to available expertise, equipment, and small incremental gains of the technologies and additional variants should also be considered.
In conclusion, this study demonstrates that while genomic testing methods have advanced, detection rates have not greatly improved, possibly due to wider inclusion criteria, and the small incremental gains of testing genes other than PKD1 and PKD2. For people at risk of ADPKD in genetically unresolved families, the accuracy of ultrasound is uncertain, and clinical communities should bear this in mind when screening for ADPKD.
Supplementary Material
sfaf187_Supplemental_Files
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chapman AB, Devuyst O, Eckardt K-U et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2015;88:17–27. 10.1038/ki.2015.5925786098 PMC 4913350 · doi ↗ · pubmed ↗
- 2Chow CL, Ong AC. Autosomal dominant polycystic kidney disease. Clin. Med 2009;9:278. 10.7861/clinmedicine.9-3-278PMC 495362219634398 · doi ↗ · pubmed ↗
- 3Choukroun G, Itakura Y, Albouze G et al. Factors influencing progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995;6:1634–42. 10.1681/ASN.V 6616348749691 · doi ↗ · pubmed ↗
- 4Mahboob M, Rout P, Leslie SW et al. Autosomal Dominant Polycystic Kidney Disease. Treasure Island, FL, USA: Stat Pearls Publishing. (date last accessed 7 August 2024), https://www.ncbi.nlm.nih.gov/books/NBK 532934/30422529 · pubmed ↗
- 5Brosnahan GM. Volume progression in polycystic kidney disease. N Engl J Med 2006;355:733–4.16914712 10.1056/NEJ Mc 061638 · doi ↗ · pubmed ↗
- 6Yu CC, Lee AF, Kohl S et al. PKD 2 founder mutation is the most common mutation of polycystic kidney disease in Taiwan. npj Genom Med 2022;7:1–11. 10.1038/s 41525-022-00309-w PMC 924987435778421 · doi ↗ · pubmed ↗
- 7Foundation P. PKD variant database. (date last accessed 7 August 2024), http://pkdb.mayo.edu/
- 8Audrezet MP, Cornec-Le Gall E, Chen JM et al. Autosomal dominant polycystic kidney disease: comprehensive mutation analysis of PKD 1 and PKD 2 in 700 unrelated patients. Hum Mutat 2012;33:1239–50. 10.1002/humu.2210322508176 · doi ↗ · pubmed ↗