An Atypical Case of CREST Syndrome With Early Complete Clinical Manifestation
Geil A Schock, Dana Simon, Ethan Weitzman, Raymond Weitzman

TL;DR
A rare case of CREST syndrome presented with all five symptoms within a year, emphasizing the need for early diagnosis and comprehensive care.
Contribution
This paper presents an atypical, rapidly progressive case of CREST syndrome with full clinical manifestation in a short timeframe.
Findings
A 44-year-old patient developed all five CREST syndrome features within one year.
The case highlights the importance of early recognition and multidisciplinary management in autoimmune diseases.
Social determinants of health were emphasized as critical factors in patient care.
Abstract
CREST syndrome, the limited cutaneous subtype of systemic sclerosis, is defined by five classic clinical features: calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia, which also forms the initialism. While diagnosis requires the presence of only three criteria, the full expression of all five is uncommon and typically develops gradually over many years. We describe the case of a 44-year-old Spanish-speaking female patient who presented to a community-based health clinic. Her initial evaluation identified the recent onset of Raynaud’s phenomenon, esophageal symptoms, sclerodactyly, and facial telangiectasia. Serology was notable for positive anti-centromere and antinuclear antibodies. One year later, she developed a painful mass on her right foot, ultimately identified as dystrophic calcification consistent with calcinosis. This case highlights a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
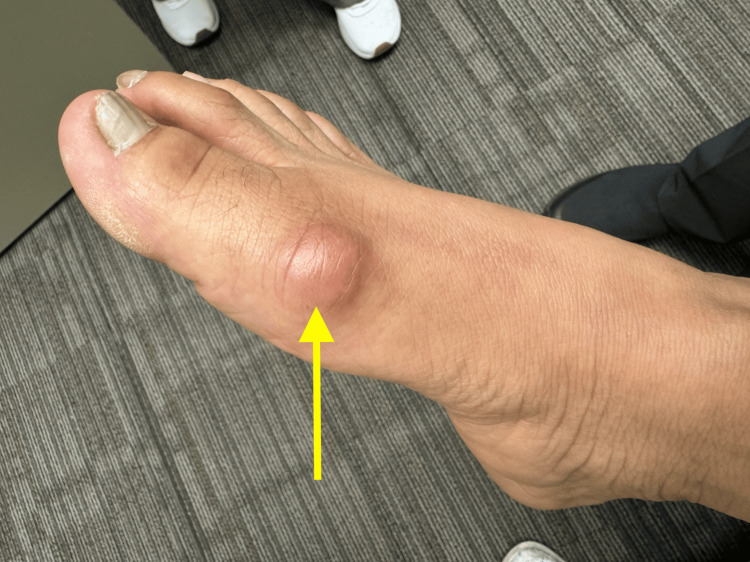 Figure 1
Figure 1| Autoantibody | Patient Value | Reference Range |
| Antinuclear (ANA) | Positive (>1:1280) | Negative (<1:80) |
| Anti-centromere | Positive | Negative |
| Anti-Scl-70 | Negative (<0.6) | <7.0 U/mL |
| Anti-RNA polymerase III | Negative (<20) | <20 Units |
| dsDNA | Negative (<0.6) | <10 IU/mL |
| Anti-Smith | Negative (<0.7) | <7.0 U/mL |
| RNP | Negative (<2.5) | <5.0 U/mL |
| Rheumatoid factor (RF) | Negative (<15) | <15 IU/mL |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSystemic Sclerosis and Related Diseases · Cutaneous Melanoma Detection and Management · Medical Imaging and Pathology Studies
Introduction
CREST syndrome, or limited cutaneous systemic sclerosis (lcSSc), is a distinct clinical subtype of the autoimmune disease systemic sclerosis (SSc) [1,2]. CREST is an initialism based on the following diagnostic criteria, the presence of three of which is required for diagnosis: calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia [1,2]. As opposed to the more severe diffuse cutaneous systemic sclerosis (dcSSc), CREST syndrome is associated with more calcium deposition in the skin (calcinosis), a higher risk of pulmonary fibrosis and pulmonary hypertension, but less widespread skin involvement. The pathophysiology of SSc is complex and characterized by an initial vascular insult that triggers platelet activation along with thrombotic and fibrinolytic cascades, ultimately leading to the development of autoimmunity and tissue fibrosis [1].
Further differentiating it from dcSSc, CREST syndrome is characteristically associated with anti-centromere antibodies as opposed to anti-Scl-70 and anti-RNA polymerase III [2,3]. Around 90% of SSc patients also present with a positive antinuclear antibody (ANA) [1,2]. SSc predominantly affects females, with a reported female-to-male ratio of approximately 5:1 and an earlier disease onset in women [1]. Global prevalence ranges from 38 to 341 cases per million persons, with annual incidence rates between 8 and 56 new cases per million [1]. The symptomatic progression of CREST syndrome typically occurs over many years, with Raynaud’s phenomenon often preceding other manifestations [1,4]. Studies have reported a mean delay of over five years between the onset of Raynaud’s phenomenon and the first non-Raynaud’s symptom in female patients with CREST syndrome [5]. Here, we describe an unusual presentation in which the full spectrum of diagnostic criteria emerged within one year of initial evaluation.
Case presentation
Initial evaluation
A 44-year-old Spanish-speaking female patient presented to a community-based clinic for medically underserved populations with the recent onset of symptoms consistent with mild, episodic Raynaud’s phenomenon, followed by gastroesophageal reflux and dysphagia. Her past medical history included hypothyroidism, type 2 diabetes mellitus, diverticulosis, and recently treated endometrial carcinoma.
On physical examination, telangiectasia was present on her face, and sclerodactyly was noted involving the proximal upper and lower extremities. Autoimmune etiology was suspected due to her widespread and specific constellation of symptoms, with SSc leading the differential.
Laboratory testing revealed high-titer positive ANA (>1:1280) and anti-centromere antibody levels. Further autoantibody testing was negative for anti-Scl-70, anti-RNA polymerase III, dsDNA, anti-Smith, rheumatoid factor, and ribonucleoprotein (RNP) antibodies (Table 1). Values for other tests, such as C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), C3 and C4 complement, total creatine kinase, and thyroid-stimulating hormone (TSH), all fell within normal limits. Due to the concern over possible interstitial lung disease and pulmonary hypertension with a likely diagnosis of CREST syndrome, an early high-resolution chest CT (HRCT) was conducted with normal findings.
Based on the patient’s clinical presentation and indicative serology results, a diagnosis of CREST syndrome was established. Given her mild symptoms and financial insecurity, conservative symptom management with over-the-counter antacid therapy for gastroesophageal reflux was recommended. She received extensive counseling on the importance of regular follow-up and pulmonary screening.
Emergence of final diagnostic criterion
The patient returned to the clinic nine months later with severe, intermittent right hallux pain. Examination revealed a mass over the right first metatarsal head without significant erythema or signs of infection (Figure 1). The remainder of her physical exam remained unchanged from previous visits. Laboratory evaluation of serum calcium, phosphorus, uric acid, and ESR was normal. Furthermore, there was no evidence of leukocytosis, and blood cultures did not isolate any pathogens. The differential diagnosis considered possible bacterial infection or inflammatory processes such as gout. Initial management with broad-spectrum antibiotics, colchicine, and anti-inflammatory medications was unsuccessful.
Subcutaneous mass over right first metatarsal head (arrow)
As her pain symptoms became persistent, the patient presented to the emergency department for evaluation. Radiography revealed a calcified lesion at the site of the palpated mass. Following referral to podiatry, the lesion was surgically excised. Histopathologic analysis of the surgical specimens identified proliferative synovial tissue, calcified fibrous tissue, and no evidence of acute or chronic inflammatory infiltrate. This finding is consistent with dystrophic calcification, the underlying pathologic process responsible for calcinosis in the setting of CREST syndrome [6]. In the broader context of her autoimmune disease, the presence of calcinosis confirms the complete expression of all five hallmark features. Due to institutional limitations within the community-based clinic where the patient was seen, original radiologic imaging and histopathologic photographs are not available for publication. However, formal reports confirming these findings were documented in the patient’s medical record.
Discussion
While the diagnosis of CREST syndrome requires at least three of five diagnostic criteria, this case is significant for the complete disease manifestation as well as the rapid progression of symptoms. A majority of patients with this condition develop the signs and symptoms over many years, particularly Raynaud’s phenomenon, which can appear years before other symptoms [1,4]. In contrast, the female patient in the current report demonstrated calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia within a one-year period. This uncommon disease trajectory underscores the importance of heightened clinical suspicion of autoimmune disorders, especially in patients with limited healthcare access.
There were multiple social factors that complicated this patient’s treatment course and clinical work-up. Because she spoke only Spanish, there was a significant communication barrier to overcome. While language interpretation services were utilized, miscommunication was likely. The patient also struggled with food insecurity and an intermittent lack of health insurance. These factors likely hindered her ability to maintain consistent follow-up, adhere to treatment, and initiate early physical therapy. Her recent history of endometrial cancer further complicated her care by diverting clinical attention away from autoimmune disease management. The compounding effects of her social determinants of health likely contributed to delayed assessment and eventual surgical excision of her lower extremity calcinosis. Now established at the community health clinic, the patient has greater access to medical care and social services.
Treatment of CREST syndrome focuses primarily on symptom management and is tailored to the patient’s specific disease manifestations. Calcinosis is typically managed with supportive care, including pain control, anti-inflammatory medications, and, in refractory cases, surgical excision [6]. First-line treatment for Raynaud’s phenomenon includes calcium channel blockers and other vasodilators [4]. Esophageal dysmotility, presenting in this patient as gastroesophageal reflux disease (GERD) symptoms, is commonly addressed with proton pump inhibitors (PPIs) [1]. For sclerodactyly, immunosuppressants such as methotrexate may help delay progressive fibrosis [7], while physical and occupational therapy play a crucial role in preserving hand function [1]. Telangiectasia is generally treated for cosmetic purposes, often with pulsed dye laser therapy [1]. A multidisciplinary approach is also essential for these patients, particularly for monitoring systemic involvement and long-term disease progression. Early and regular screening for pulmonary complications, such as interstitial lung disease and pulmonary hypertension, with HRCT is critical even in asymptomatic patients [1].
In this patient’s case, long-term success will depend on continued engagement with her multidisciplinary care team. Support from primary care, rheumatology, pulmonology, and social services will be essential in addressing her condition and barriers to care.
Conclusions
This case demonstrates a rare and complete presentation of CREST syndrome, identified by all five diagnostic criteria (calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia) as well as positive serology (ANA, anti-centromere). This case was further atypical due to her rapid disease progression within approximately 12 months, while symptoms generally progress over multiple years. Given this unusual course, clinicians should consider early serologic testing for patients presenting with Raynaud’s phenomenon and overlapping systemic disease manifestations.
Equally important are the social factors that significantly impacted this patient’s healthcare, both in terms of access and treatment compliance. She faced multiple barriers, including language differences, a lack of consistent health insurance, and food insecurity. These socioeconomic challenges are not uncommon in chronic disease patients, necessitating recognition to improve disease morbidity and overall burden.
In summary, this case presentation showcases a rare, full expression of CREST syndrome with rapid disease progression. It reinforces the importance of early symptom identification, serologic testing, pulmonary screening, and a multidisciplinary approach to improve outcomes in underserved populations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Systemic sclerosis (scleroderma)Stat Pearls [Internet] Adigun R Goyal A Hariz A Treasure Island (FL)Stat Pearls Publishing 2025 https://www.ncbi.nlm.nih.gov/books/NBK 430875/28613625 · pubmed ↗
- 2Diagnosis and classification of systemic sclerosis Clin Rev Allergy Immunol Hachulla E Launay D 78834020112014318210.1007/s 12016-010-8198-y · doi ↗ · pubmed ↗
- 3Autoantibodies in systemic sclerosis: unanswered questions Front Immunol Kayser C Fritzler MJ 167620152592683310.3389/fimmu.2015.00167 PMC 4397862 · doi ↗ · pubmed ↗
- 4Raynaud's phenomenon with focus on systemic sclerosis J Clin Med Maciejewska M Sikora M Maciejewski C Alda-Malicka R Czuwara J Rudnicka L 24901120223556661410.3390/jcm 11092490 PMC 9105786 · doi ↗ · pubmed ↗
- 5Epidemiology of systemic sclerosis in the UK: an analysis of the Clinical Practice Research Datalink Rheumatology (Oxford) Pauling JD Mc Grogan A Snowball J Mc Hugh NJ 268826966020213321250410.1093/rheumatology/keaa 680 · doi ↗ · pubmed ↗
- 6Calcinosis cutis Stat Pearls [Internet] Le C Bedocs PM Treasure Island (FL)Stat Pearls Publishing 2025 https://www.ncbi.nlm.nih.gov/books/NBK 448127/28846311 · pubmed ↗
- 7Emerging treatments for scleroderma/systemic sclerosis Fac Rev Zhu JL Black SM Chen HW Jacobe HT 431020213413165310.12703/r/10-43PMC 8170563 · doi ↗ · pubmed ↗