Characterization of Staphylococcus argenteus in Christchurch, New Zealand, and comparison to global strains
Trevor Anderson, Hui Wang, Michael Harrington, Julia C. Howard, Erik Otte

TL;DR
This study characterizes five Staphylococcus argenteus strains from New Zealand and compares their genomic features to global strains.
Contribution
The paper provides new genomic data on S. argenteus strains from Christchurch, New Zealand.
Findings
Four isolates belonged to ST2250 and one to ST2793, showing genetic diversity.
Three isolates had SCCmec type IV 2Bc, indicating beta-lactam resistance.
No TSST-1 or PVL genes were detected, despite shared virulence genes with global isolates.
Abstract
Staphylococcus argenteus (SARG) was discovered in 2009 as part of the Staphylococcus aureus (SAUR) complex and has been documented from various locations worldwide. In this article, we describe the genomic features of five strains of SARG found in Christchurch, New Zealand. Isolates were first detected in 2019 using MALDI-TOF identification, and their identities were confirmed using whole-genome sequencing. Genomic features, including antimicrobial resistance markers and virulence factors, were compared with other SARG sequences in the NCBI GenBank and well-characterized features in SAUR. Four isolates belonged to ST2250 and one isolate to ST2793. Phylogenetic analysis based on core genome analysis revealed that all five isolates were phylogenetically distinct, with four isolates clustering in the ST2250 clade. Three isolates contained staphylococcal cassette chromosome mec (SCCmec)…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Fig. 1
Fig. 1 Fig. 2
Fig. 2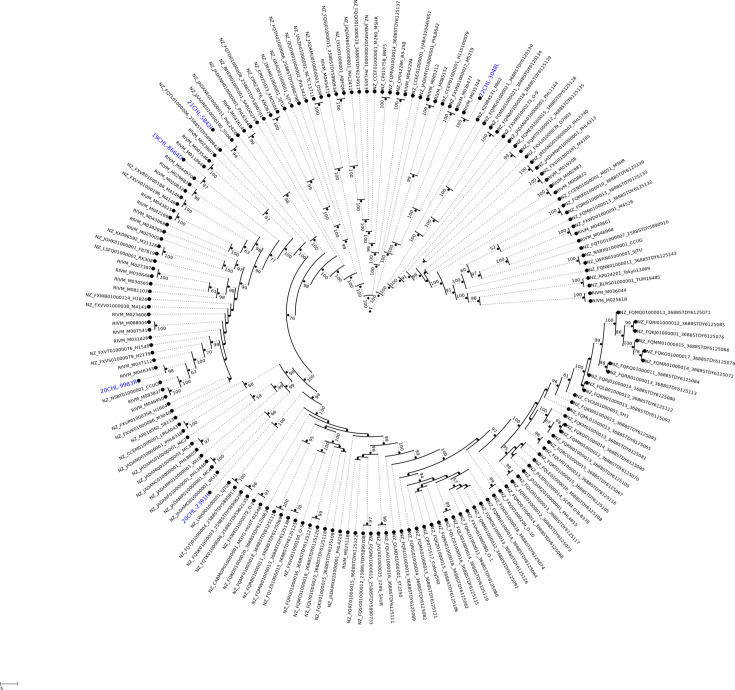 Fig. 3
Fig. 3| Sample | Contig | Genome size (Mbp) | N50 (bp) | GC (mol%) | CDS | Kraken 2 ID |
|---|---|---|---|---|---|---|
| 19CHL-8664D | 34 | 2.8 | 297,335 | 32.29 | 2,667 | SARG |
| 20CHL-2391H | 38 | 2.76 | 388,839 | 32.26 | 2,597 | SARG |
| 20CHL-9983R | 29 | 2.8 | 349,638 | 32.27 | 2,661 | SARG |
| 21CHL-5842I | 22 | 2.72 | 487,332 | 32.31 | 2,566 | SARG |
| 22CHL-3048L | 91 | 2.75 | 140,563 | 32.26 | 2,569 | SARG |
| Antimicrobial (µg) | 19CHL-8664D | 20CHL-9983R | 20CHL-2391H | 21CHL-5842I | 22CHL-3048L* |
|---|---|---|---|---|---|
| Penicillin (10U) | R (10) [ | R (10) ( | S (38) | S (38) | R ( |
| Flucloxacillin (1) | R (11) [ | R (12) ( | S (22) | S (22) | R ( |
| Gentamicin (10) | S (22) | S (21) | S (22) | S (23) | S |
| Erythromycin (15) | S (23) | S (25) | S (26) | S (24) | S |
| Clindamycin (2) | S (23) | S (24) | S (28) | S (25) | S |
| Co-trimoxazole (25) | S (19) | S (17) | S (27) | S (30) | S |
| Tetracycline (30) | S (30) | S (27) | S (30) | S (27) | S |
| Fosfomycin (200) | (20) ( | (25) ( | (22) ( | (20) ( | Not tested |
| Norfloxacin (10)† | S (23) | S (24) | S (25) | S (24) | S |
| Clindamycin induction | Not detected | Not detected | Not detected | Not detected | Not detected |
| Trimethoprim | Not tested ( | Not tested ( | Not tested | Not tested | Not tested ( |
| Antimicrobial class | Gene | NZ (5) | Global (175) |
|---|---|---|---|
| Aminoglycoside |
| 0 | 2 |
|
| 0 | 1 | |
|
| 0 | 55 | |
| Beta-lactam |
| 0 | 2 |
|
| 3 | 111 | |
|
| 3 | 58 | |
| Bleomycin |
| 0 | 2 |
| Fosfomycin |
| 4 | 152 |
| Fusidic acid |
| 0 | 1 |
| Lincomycin-Clindamycin | 0 | 1 | |
| Macrolide | 0 | 2 | |
| Macrolide-Streptogramin B | 0 | 1 | |
| Tetracycline |
| 5 | 175 |
| 0 | 8 | ||
| 0 | 54 | ||
| Trimethoprim |
| 0 | 1 |
|
| 3 | 41 |
| Gene | Virulence factor | 19CHL-8664D | 20CHL-9983R | 20CHL-2391H | 21CHL-5842I | 22CHL-3048L |
|---|---|---|---|---|---|---|
|
| Adenosine synthase A | + | + | + | + | + |
|
| Aureolysin | + | + | + | + | + |
| Capsular polysaccharide | + | + | + | + | (+) | |
| Clumping factor | +/+ | +/+ | +/+ | +/+ | +/+ | |
|
| Cytoplasmic protease | + | + | + | + | + |
|
| Fibrinogen-binding protein | + | + | + | + | + |
| Type VII protein secretion system | +/+/− | +/+/− | +/+/− | +/+/− | +/+/+ | |
| Type VII protein secretion system | +/+/+ | +/+/+ | +/+/+ | +/+/+ | +/+/+ | |
| Cell membrane protein | +/− | +/− | +/− | +/− | +/+ | |
| Fibronectin (Fn)-binding protein | +/+ | +/+ | +/+ | +/+ | +/+ | |
|
| Glycerol ester hydrolase | + | + | + | + | + |
| Gamma haemolysin | +/+/+ | +/+/+ | +/+/+ | +/+/+ | +/+/+ | |
|
| Alpha haemolysin | + | + | + | + | + |
|
| Hyaluronate lyase | + | + | + | + | + |
| Intercellular adhesion genes | +/+ | +/+ | +/+ | +/+ | +/+ | |
| Iron-regulated surface proteins | + | + | + | + | + | |
|
| Lipase | + | + | + | + | + |
| Haemolysin | +/− | +/− | +/− | +/− | +/− | |
|
| Staphylokinase | + | + | + | − | − |
|
| IgG-binding protein | + | + | + | + | + |
|
| Staphylococcus complement inhibitor | + | + | + | − | + |
| Serine-aspartate repeat-containing protein | +/−/− | +/+/+ | +/+/+ | −/+/+ | +/+/+ | |
|
| Staphylococcal protein A | + | + | + | + | + |
| Serine protease | +/+/+ | +/+/+ | +/+/+ | +/+/+ | +/+/+ | |
|
| von Willebrand factor-binding protein | + | + | + | + | − |
| Pathogenic function | Gene | NZ (5) | Global (175) |
|---|---|---|---|
| Adhesion and biofilm |
| 1 | 9 |
| Adhesion and biofilm |
| 3 | 56 |
| Adhesion and biofilm |
| 0 | 5 |
| Adhesion and biofilm |
| 5 | 161 |
| Adhesion and biofilm |
| 3 | 154 |
| Adhesion and biofilm |
| 5 | 175 |
| Adhesion and biofilm |
| 5 | 142 |
| Adhesion and biofilm |
| 4 | 141 |
| Adhesion and biofilm |
| 4 | 138 |
| Capsule biosynthesis |
| 5 | 174 |
| 5 | 175 | ||
|
| 4 | 165 | |
|
| 4 | 168 | |
|
| 4 | 167 | |
| Haemolysin |
| 5 | 175 |
| Haemolysin |
| 5 | 173 |
| Haemolysin |
| 5 | 175 |
| Immune evasion |
| 5 | 175 |
| Immune evasion |
| 5 | 175 |
| Immune evasion |
| 3 | 108 |
| Immune evasion |
| 5 | 175 |
| Immune evasion |
| 4 | 140 |
| Iron acquisition |
| 5 | 175 |
| Protease |
| 5 | 175 |
| Protease |
| 5 | 175 |
| Protease |
| 5 | 175 |
| Secretion system |
| 5 | 175 |
| Secretion system |
| 1 | 41 |
| Secretion system |
| 5 | 175 |
| Secretion system |
| 5 | 175 |
| Secretion system |
| 1 | 41 |
| Secretion system |
| 5 | 175 |
| Secretion system |
| 1 | 41 |
| Tissue-degrading enzyme |
| 5 | 173 |
| Toxin |
| 0 | 1 |
| Toxin |
| 5 | 134 |
| Toxin |
| 0 | 7 |
| Toxin |
| 0 | 11 |
| Toxin |
| 0 | 2 |
| Toxin |
| 0 | 19 |
| Toxin |
| 5 | 174 |
| Toxin |
| 5 | 175 |
| Toxin |
| 0 | 2 |
- —http://dx.doi.org/10.13039/501100006571 Canterbury District Health Board
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntimicrobial Resistance in Staphylococcus · Bacterial biofilms and quorum sensing · Bacterial Identification and Susceptibility Testing
Data Summary
All raw MiSeq sequence data and assemblies have been added to GenBank, accession numbers: PRJNA957538–SAMN34257150 (19CHL-88664D), 20CHL-2391H (SAMN34257151), 20CHL-9983R (SAMN34257152), 21CHL-5842I (SAMN34257153) and 22CHL-3048L (SAMN34257154).
Introduction
Staphylococcus argenteus (SARG) is a member of the Staphylococcus aureus (SAUR) complex and was first discovered in tropical northern Australia as a unique lineage within SAUR using MLST and spa typing. Initially referred to as clonal complex 75, SARG has been subsequently found in other populations around the world [13]. A recent article by Witteveen et al. [4] has shown evidence of SARG circulating in the Netherlands since at least 2008 [4]. Additionally, isolates have been described in animals in small numbers: from a cow in Malaysia, a pig in China, a gorilla in Gabon [5], a dog in the Netherlands [6] and in retail chicken samples in China and Japan, as well as in slaughterhouses in Japan [7].
There are no phenotypic features that can reliably identify SARG from SAUR other than the lack of staphyloxanthin (golden pigment) production [48]. Genomic analysis has found that SARG lacks the carotenoid biosynthetic operon crtOPQMN. MALDI-TOF MS may differentiate SARG from SAUR [9], but molecular assays such as nuc gene PCR [10] or DNA sequencing of various genes are required for confirmation [1113]. SARG strains encoding the mecA gene within the SCCmec region may also be falsely identified as methicillin-resistant Staphylococcus aureus (MRSA) when screening samples using phenotypic testing. Subsequently, SARG isolates are likely commonly misidentified, partly explaining why they are rarely detected in the clinical laboratory. This was demonstrated by a study from Thailand that retrospectively examined 246 consecutively collected SAUR isolates and found that 4.1% have matching MLST arC and pta allele profiles associated with SARG as determined by phylogenetic clustering [2].
SARG has been isolated from a range of clinical sites. Although it was initially considered to be less pathogenic than SAUR, there are increasing reports of serious invasive disease including bacteraemia and bone and joint infections caused by the organism [1415]. It has been suggested that the lower pathogenicity of SARG could be due to a lack of staphyloxanthin production, which protects against oxidative stress [12]. SARG strains have been described as harbouring pathogenic genes such as Panton–Valentine leukocidin (PVL) and toxic shock syndrome toxin-1 (TSST-1) [1618], and an invasive infection study from Thailand found that SARG has similar rates of morbidity and mortality as methicillin-susceptible SAUR [16].
Plasmids play a crucial role in bacterial evolution and often carry resistance genes, virulence factors and metabolic capabilities, such as heavy metal resistance. Horizontal gene transfer is an important mechanism for plasmid exchange and has been documented between Staphylococcus spp. [1921].
In this article, we describe the genomic features of five SARG isolates from New Zealand (NZ), comparing genomic characteristics including antimicrobial resistance genes and pathogenic markers, with global SARG isolates available in the NCBI GenBank. SARG is part of the SAUR complex and closely related to SAUR. Therefore, we also compared key pathogenic markers and other genomic features with those of this well-characterized pathogen.
Methods
This study involved five identified routinely collected clinical isolates of SARG received between 2019 and 2022, which underwent whole genome sequencing (WGS) to confirm their identification. As per usual laboratory processes for routine MRSA screening, isolates from swab samples (nose, groin and other skin sites) were recovered from SAUR selective enrichment broth, which was sub-cultured onto BBL™ CHROMagar™ MRSA II agar (Fort Richards, Auckland, NZ). All media were incubated at 35 °C for 24 h in an ambient atmosphere. Pink colour colonies from chromogenic agar or colonies resembling SAUR from 5% sheep blood agar (SBA) were identified as SAUR/SARG using Bruker Microflex LT (Bruker Daltonics, Bremen, Germany). A blood culture isolate was plated from a BD BACTEC Plus aerobic bottle inoculated onto 5% SBA. MALDI-TOF MS was performed on pure culture of the isolate by inoculation onto the MALDI target plate using a wooden applicator, followed by the application of 1 µl formic acid, which was air dried before applying 1 µl α-cyano-4-hydroxycinnamic acid matrix; results were analysed with a database containing 6,903 Mass Spectral Profiles (MSP). Isolates were sub-cultured onto 5% SBA and stored at −80 °C in cryo-preserved vials.
Antimicrobial susceptibility testing
Isolates were tested using European Committee on Antimicrobial Susceptibility Testing (EUCAST) guidelines and categorized as susceptible (S), susceptible increased exposure (I) or resistant (R) based on v11.0 breakpoints. One isolate was non-viable from storage, but initial sensitivity results using the BD Phoenix PMIC-84 panel were available. Clindamycin-inducible erm-mediated resistance was screened using the D-zone agar diffusion test with erythromycin [22].
Molecular analysis
Nucleic acid was extracted using the QIAamp UPC pathogen mini kit (Qiagen, Auckland, NZ) using mechanical pre-lysis and spin protocol as recommended in the handbook (v04/2021). DNA was quantitated for WGS using Qubit fluorometer (Thermo Fisher Scientific) with the dsDNA broad range kit. WGS DNA library preparation was performed using the Illumina DNA Flex library kit (Illumina, Australia). Libraries were sequenced using MiSeq (Illumina) with micro flow-cell, reagent kit v2 (300 cycles) according to the manufacturer’s instructions. Bioinformatic analysis was performed with the following software with default settings, unless stated otherwise. FASTQ paired-end reads were quality trimmed and assembled into contiguous (contigs) sequences using Shovill v0.9.0 (https://github.com/tseemann/shovill). Genome assembly quality and accuracy were checked using CheckM2 [23]. Genomic analysis was performed with the following software: Kraken 2 [24], AMRFinder [25], MLST (https://github.com/tseemann/mlst) and Abricate (https://github.com/tseemann/abricate) with MEGARes database (6,635, 3 December 2024). SCCmec typing and plasmid (PlasmidFinder) typing were performed via the Center for Genomic Epidemiology online server http://www.genomicepidemiology.org/services/ (accessed 14 February 2022). Additionally, 227 global SARG sequences from RefSeq (NCBI) were downloaded on 29 November 2024 for comparative analysis. Dereplicator software (https://github.com/rrwick/Assembly-Dereplicator) was used to filter 52 redundant sequences based on a 0.001 distance score. One hundred and eighty FASTA sequence files were annotated using Prokka [26], and the GFF3 files were then analysed by the Panaroo program v1.5.1 [27] to create a core genome. Phylogenetic analysis was performed on the core genome SNP alignment with Gubbins v3.3.1 [28] recombination filtering and IQ-TREE v2.3.6 [29] tree construction using maximum-likelihood modelling with 1,000 bootstrap calculations. Plasmid contigs were reoriented around the rep gene using dnaapler v1.1.0 (https://github.com/gbouras13/dnaapler) and annotated using bakta v1.1.10 software [30]. The annotated sequences were then aligned using the MAFFT plugin in Geneious Prime (2025.0.2). Annotated genomes were viewed for gene synteny using Geneious Prime.
Whole-genome data from this publication can be found in the NCBI GenBank under BioProject PRJNA957538. BioSample numbers are as follows: SAMN34257150 (19CHL-88664D), SAMN34257151 (20CHL-2391H), SAMN34257152 (20CHL-9983R), SAMN34257153 (21CHL-5842I) and SAMN34257154 (22CHL-3048L).
Results
Four SARG were isolated from skin sites and one from blood culture. Isolates grown on 5% SBA typically lacked the characteristic golden pigment (staphyloxanthin), which is normally present in SAUR colonies. MALDI-TOF MS profiles failed to differentiate SARG from SAUR with a best match score of ≥1.9 but highlighted potential isolates for further analysis. Identification as SARG was confirmed using WGS with Kraken 2 and blastn (https://blast.ncbi.nlm.nih.gov/) of the thermonuclease (nuc) gene. The analysis of the annotated sequences found no complete carotenoid biosynthetic operon (crtP, crtQ, crtM or crtN genes), which is consistent with the phenotypic appearance. blastn analysis of the nuc gene, which is ~675 bp, found matching nt identities of 98–100% to other SARG sequences but only 93% match with SAUR and Staphylococcus schweitzeri.
The FASTQ files were assembled using the Shovill pipeline that uses the Spades assembler [31], and genome assembly checkm2 metrics are available in Table 1.
Antimicrobial susceptibility
The NZ SARG isolates were susceptible to a wide range of antimicrobials with notable resistance to penicillin and flucloxacillin. The whole-genome analysis also detected blaZ and mecA genes which encode beta-lactam resistance and fosB encoding fosfomycin resistance as seen in Table 2. No EUCAST interpretative guidelines are available for fosfomycin disc susceptibility testing, but the presence of fosB has been described in SAUR as a mechanism of resistance [32]. The three methicillin-resistant isolates had SCCmec mobile genetic element type IVc (2B) containing IS6 family transposase (IS431mec) and mecA, mecI and mecR genes. The analysis of 175 global strains from GenBank showed that blaZ is the predominant mechanism for penicillin resistance, present in 111 (63.4%) isolates (Table 3). The other beta-lactam resistance mechanism detected was altered penicillin-binding protein (PBP) 2a encoded by the mecA gene found in 58 (33.1%) isolates. Other significant resistance markers included fosB in 152 (86.9%) isolates, aph(3′)-IIIa (aminoglycoside resistance) in 55 (31.4%) and dfrG (trimethoprim resistance) in 41 (23.4%) (see Table 3). The five NZ isolates were relatively susceptible with no macrolide or lincosamide (inducible or constitutive), fluoroquinolone or tetracycline resistance by phenotypic or genotypic testing. Other resistance mechanisms detected included multidrug and toxic compound extrusion (MATE) and major facilitator superfamily (MFS) efflux pumps, such as tetracycline resistance (TET38).
Virulence factors
The analysis of SARG virulence factor profiles revealed that many of the same virulence factors present in SAUR were also found in the NZ SARG isolates, summarized in Table 4. Most virulence factors were common to all isolates with variations such as ESAT-6 secretion system (esaC), ESAT-6 cell membrane secretion system (esxB), staphylokinase (sak), Staphylococcus complement inhibitor (scn), serine-aspartate repeat-containing proteins (srdC/D/E) and von Willebrand factor-binding protein (vWbp). Isolate 22CHL-3048L was missing genes encoding the capsular polysaccharide cap8H-K and clustered with seven global isolates also without these cap8 genes (see Fig. 1). Comparison of virulence factors of 175 global strains from the GenBank showed that they shared the same virulence factors, notably those responsible for capsular production, haemolysin, toxin secretion and fibrinogen factors (see Table 5).
Phylogenetic tree with mapped genomic features. The maximum-likelihood phylogenetic tree from Fig. 3 with genomic features such as MLST, antimicrobial and virulence factors. Data was generated using Abricate software with the MEGARes and VFDB databases, along with PlasmidFinder, SCCmecFinder and AMRfinder.
Genome variation of NZ SARG. This figure shows the Panaroo-constructed pangenome with the core genome containing 81.3% gene content. The accessory genome includes unidentified coding regions, pathogenic factors and mobile genetic elements such as plasmids and SCCmec.
Phylogenetic comparison of NZ and global SARG strains. The distribution of global SARG strains, with NZ strains highlighted, is shown on a mid-point rooted maximum-likelihood phylogenetic tree, created by IQ-TREE (1,000 bootstrap replicates) from the Panaroo core gene SNP alignment. The circular format clearly shows the tree structure and strong branch support values.
Plasmid analysis
Plasmid typing was performed by PlasmidFinder tool, and representative plasmid sequence(s) were downloaded from https://www.ccb.uni-saarland.de/plsdb [33]. SARG penicillin-resistant isolates 19CHL-8664D and 20CHL-9983R contained replication loci (rep) types rep16_1_CDS8(pSAS) and rep5a_1_repSAP001(pN315), and isolate 22CHL-3048L contained rep20_3_rep(pTW20). Rep16 (size range 20,622 to 21,326 bp), rep20 (size range 20,403 to 33,660 bp) and rep5a (size range 19,758 to 59,392 bp) contain the blaZ, blaI and blaR1 genes. Penicillin-susceptible isolate 20CHL-2391H had rep13 type (size range 2,779 to 3,145 bp) with no blaZ gene, and isolate 21CHL-5842I had no rep types detected. Analysis of the global strains found that 144 (82.3%) had a replicon locus detected, with the most common types being 119 (68%) rep5 and 127 (72.6%) rep16, with 118 (67.4%) isolates having both replicon types. The same replicon types found in SARG are also in SAUR, indicating a potential exchange of mobile genetic elements [34]. Plasmids rep5a and rep20 also contain the cadD gene, which codes for the cadmium efflux system and high-level resistance to this heavy metal [35]. The blaZ plasmids for isolates 19CHL-8664D (20,755 bp contig) and 20CHL-9983R (20,736 bp contig) were aligned against plasmid database (PLSDB) (https://ccb-microbe.cs.uni-saarland.de/plsdb2025) sequences and found similar blaZ containing plasmids around 20 kb with rep_cluster_1733 and rep_cluster_2214. The 19CHL-8664D and 20CHL-9983R plasmids showed near identical gene synteny, except for 19CHL-8664D that had a 111 bp insert in the nifR gene. Isolate 22CHL-3084L also had a blaZ gene-containing plasmid (20,108 bp contig), which had 99.9% blastn alignment to plasmid CP096113.1 (25,332 bp) with PLSDB rep_cluster_1215. The 22CHL-3084L plasmid fragment has a 5,225 bp region missing that contains hypothetical proteins, transposons and origin of transfer (oriT). Apart from this missing region, the plasmid shows identical gene synteny to the CP096113.1 plasmid. Isolate 20CHL-2391H plasmid fragment (4,788 bp) showed partial alignment on blastn (1,898/1,953 bp) to Staphylococcus warneri p12 plasmid (2,140 bp) (AB125342.1) and contains efflux gene qacC.
Molecular typing and phylogenetic analysis
The NZ isolates were sequence typed as ST2250 (n=4) and ST2793 (n=1), with ST2250 being the most common sequence type (ST) group among the global SARG strains. The analysis of the 175 global strains showed the following ST types: 122 (69.7%) ST2250, 22 (12.6%) ST1223, 8 (4.6%) ST2198, 6 (3.4%) ST2793, 5 (2.9%) ST2854, 3 (1.7%) ST1850, 2 (1.1%) ST5978, 1 (0.6%) ST3261, 1 (0.6%) ST6111 and 6 (3.4%) unknown MLSTs, which have not been uploaded to the PubMLST database.
Pangenome of the NZ isolates constructed using the Panaroo program identified 2,830 gene families, with the core genome containing 2,274 (81.3%) at 95–100% identity. Although four isolates share the same MLST profile, they are genetically distinct based on core genome analysis and accessory gene content (see Fig. 2). The accessory genome contained 556 CDSs that consisted mainly of 388 (69.8%) hypothetical proteins. Additionally, the accessory genome included SCCmec elements such as insertion sequences, recombinases, heavy metal resistance (e.g. cadC for cadmium resistance) and antibiotic resistance (e.g. mecA). Other notable genes found included accessory genes and plasmid-associated genes, such as phage tail proteins, phage major capsid protein and plasmid recombination enzymes. Additionally, a Panaroo pangenome was constructed including 175 global strains. The pangenome identified 3,684 gene families across all 180 strains, with the core genome containing 2,196 (61.1%) genes at 95–100% identity. The maximum-likelihood phylogenetic tree strongly supports the presence of six to seven clades, with the NZ SARG strains distributed across two clades (see Figs 1 and 3). The MLST types align within these clades with a few exceptions. The largest clade, which is predominantly ST2250, contains four of the NZ isolates. Within this ST2250 clade, distinct genomic patterns define various subclades. For example, one subclade includes aph(3′)-IIIa (aminoglycoside resistance), blaZ (beta-lactam resistance), tetL (tetracycline resistance) genes, sak (staphylokinase) and replicon types rep5a and rep16. Another subclade within the ST2250 clade contains strains carrying dfrG (trimethoprim resistance) and SCCmec (mecA, beta-lactam resistance) and the same replicon types (rep5a and rep16). Another notable clade contains the ST2793 strains, which contain isolates lacking some of their cap8 genes, dfrG gene and SCCmec, but also harbouring replicon types 20/21.
Discussion
SARG is a newly described species within the SAUR complex and has been found distributed around the world causing a similar spectrum of human infections as SAUR. Phenotypic identification is difficult and remains the main challenge to many diagnostic laboratories, with reliance on colony appearance, biochemical tests such as Pyrrolidonyl aminopeptidase test, Staphylococcus Protein A, tube coagulase or MALDI-TOF MS mass spectrum profiles for identification [18]. The inclusion of SARG MSP into the Bruker database has made scientists aware of potential isolates, and mass peak profiles have been documented to differentiate the SAUR complex [9]. However, SARG still requires additional confirmation [9]. As no biochemical tests are available for definitive identification, molecular techniques such as nuclease (nuc) gene PCR are required [183637]. It is for this reason that the true incidence and disease spectrum of SARG has remained hidden due to misidentification as SAUR. Sanger sequencing of the nuclease gene is the most reliable means of identification, but this technique is not widely available in diagnostic laboratories. Likewise, applications using WGS are mostly limited to research or reference laboratories and not available to most diagnostic laboratories due to the cost and bioinformatic requirements.
SARG lack the dehydrosqualene synthase-encoding ctrM/N/P/Q genes required for the biosynthesis of staphyloxanthin, a pigment which provides oxidative protection from biological peroxides found in neutrophils and a major virulence factor in SAUR. WGS sequence analysis of our SARG strains showed no complete staphyloxanthin operon present, which is consistent with the selected global SARG sequences. Although SARG lacks this pathogenic feature, it is still a cause of disease in immunocompetent hosts with many other virulence genes associated with immune evasion, exotoxin production and cell adhesion.
A comparison of our NZ isolates with 175 global SARG genomes from the NCBI RefSeq nt database showed that many of the same SAUR virulence factors that are important for organism adaptation to different environments are also found in SARG. These virulence factors include type 8 polysaccharide capsule, host immune modification, haemolysis production (alpha and gamma), fibronectin-binding proteins, iron binding, immunoglobulin binding and proteinases. None of the NZ isolates had cytolytic toxin PVL genes lukF/S or tsst-1 gene, but the analysis of the global sequences found seven isolates with lukF/lukS-PV and two isolates with tsst-1 genes, indicating that they are rarely seen in SARG. Isolate 22CHL-3048L and seven global isolates (ST2793 clade) have a deletion of four cap8 genes (capH, capI, capJ and capK), which are part of the operon encoding the capsular polysaccharide production and is a major pathogenic factor in SAUR. The capsular polysaccharide is important in phagocytosis evasion and is thought to help in antimicrobial resistance. The loss of cap8 genes would have a detrimental effect on capsular production such as truncated or non-functional capsule production. Non-encapsulated SAUR isolated from bovine mastitis was not seen as less pathogenic but caused persistent infections [38]. This chronic mastitis was caused by the ability of non-encapsulated strains to adhere more effectively to host cells, become internalized and evade immune responses. Additionally, a study comparing acute and chronic osteomyelitis found a higher prevalence of non-encapsulated SAUR with chronic infection [39].
Antimicrobial resistance screening found that genes for fosfomycin, beta-lactams, trimethoprim and various efflux pump mechanisms are associated with fluoroquinolone or tetracycline resistance. Four out of the five NZ isolates possess the chromosomal fosB gene encoding fosfomycin resistance [32]. However, there are no EUCAST interpretive breakpoints for disc testing, but MICs are available for the confirmation of phenotypic resistance. The analysis of the 175 global strains found 152 (86.9%) isolates with the fosB gene. The fosB gene can be carried by mobile genetic elements such as plasmids where it is over-expressed [40] but is most commonly found on the chromosome [5]. The fosB gene has also been found in other Staphylococcus species such as Staphylococcus capitis, S. warneri and Staphylococcus saprophyticus [41].
Beta-lactam resistance was found to occur by two mechanisms: (i) penicillin resistance caused by class A beta-lactamase encoded by the blaZ gene and (ii) altered PBP2a encoded by the mecA gene. The mecA gene was found in isolates 19CHL-8664D, 20CHL-9983R and 22CHL-3048L, which all had SCCmec type IV 2Bc mobile genetic element. Isolates 19CHL-8664D and 20CHL-9983R were obtained from MRSA screening samples and 22CHL-3048L from blood culture; they were originally misidentified as MRSA. These isolates were phenotypically uncharacteristic of SAUR, with colony appearance lacking pigment on 5% SBA and altered colour on chromo-agar. Further investigation of the MALDI-TOF MSP indicated a possible match to SARG, prompting further analysis with WGS. Comparison with the 175 global sequences for beta-lactam resistance found blaZ enzyme is the most common mechanism of beta-lactam resistance with 111 (63.4%) out of 175 global isolates compared with 58 (33.1%) strains with mecA. Treatment of infections with methicillin-resistant SARG should be the same as for MRSA [1842]. It was observed that the dfrG gene was only found in the SCCmec-containing isolates and 41/58 (70.7%) mecA-positive global isolates. No NZ isolates possessed the tetL gene, which is associated with tetracycline resistance, but it was found in 54/175 (30.9%) of global SARG isolates. No resistance mutations were detected in the gyrA or parC genes for fluoroquinolones or ileS gene for mupirocin, using the AMRFinder program. Also, a recent study described a SARG strain with the mupirocin resistance gene mupA encoded on a plasmid [43], but this was not detected in any of the five NZ strains or 175 global sequences obtained from the RefSeq database.
Plasmid analysis found near-identical or structurally similar plasmids in SAUR and SARG sequences in PLSDB. The blaZ-harbouring plasmids were ~20 kbp in size and exhibited the same gene synteny as global strains. This suggests that blaZ is mobilized on plasmids and transferred between Staphylococcus species [44]. Replicon typing of the blaZ plasmids was consistent between the NZ isolates and their matching PLSDB plasmids. The replicon types rep5a and rep16 were detected from the same plasmid, suggesting the presence of two replication initiator protein genes. However, this is highly unlikely, as such a rare occurrence would create conflicts in the replication machinery of the cell. Isolate 20CHL-2391H (rep13) contained a 4,788 bp plasmid fragment encoding a quaternary ammonium efflux pump (qacC). A PLSDB search found no matching plasmids, suggesting that this could be a novel plasmid. However, due to the limitations of short-read sequencing data, further plasmid analysis is difficult [45] and would require long-read sequencing (e.g. Nanopore MinION) data for accurate plasmid construct and gene feature characterization. Only isolate 21CHL-5842I did not contain a plasmid as determined by replicon typing, but the presence of an unidentified replicon type cannot be excluded and long-read sequencing would be needed to confirm.
A total of nine MLST types were identified among the 175 SARG sequences from RefSeq, with ST2250 (122/175) being the most common. The NZ isolates belong to MLST types ST2250 or ST2793. While MLST typing showed some correlation with the phylogenetic clades, its resolution power is limited due to the conserved nature of the selected genes. Core genome SNP-based analysis revealed that the NZ strains are distributed across different clades, suggesting strain diversity and multiple introductions via global dissemination of SARG. The phylogenetic tree from this study was comparable to the findings by Goswami et al. [5], which identified three distinct clades with five subclades. Clade A, the largest, contained three subclades that were predominately composed of MLST ST2250. The subclade A1 included the dfrG (trimethoprim resistance), mecA (beta-lactam resistance), which included 19CHL-8664D and 20CHL-9983R. Another notable clade is the clade C (ST2793), which contains isolate 22CHL-3084L. This clade consists of isolates lacking some of their cap8 genes, but also harbouring dfrG, mecA and rep20 plasmid.
Four of the five NZ isolates were found from skin sites and one from a case of bacteraemia. The skin site isolates were all detected from MRSA screening samples, and two of the SARG isolates were methicillin resistant and indistinguishable from MRSA. The bloodstream isolate, which was methicillin resistant, underscores the pathogenic potential of this emerging organism and adds to the growing number of documented infections in the literature. The development of antimicrobial resistance due to the presence of the SCCmec element and plasmids, similar to SAUR, highlights the importance of accurately identifying and characterizing this unique species.
Conclusion
In this article, we demonstrated the close relationship between SARG found in NZ and global strains, highlighting the worldwide spread of this emerging species. SARG is now recognized as a human pathogen, sharing many of the key virulence factors found in SAUR. The presence of the SCCmec and other antimicrobial resistance factors has important implications for treatment strategies, similar to those for SAUR. Since documented cases of SARG remain limited, further global investigations are needed to reduce sampling bias in the data and improve our understanding of this organism. This publication is the first documented occurrence of SARG in NZ.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ng JWS Holt DC Lilliebridge RA Stephens AJ Huygens F et al Phylogenetically distinct Staphylococcus aureus lineage prevalent among indigenous communities in northern Australia J Clin Microbiol 2009472295230010.1128/JCM.00122-0919420161 PMC 2708510 · doi ↗ · pubmed ↗
- 2Thaipadungpanit J Amornchai P Nickerson EK Wongsuvan G Wuthiekanun V et al Clinical and molecular epidemiology of Staphylococcus argenteus infections in Thailand J Clin Microbiol 2015531005100810.1128/JCM.03049-1425568440 PMC 4390622 · doi ↗ · pubmed ↗
- 3Hansen TA Bartels MD Høgh SV Dons LE Pedersen M et al Whole genome sequencing of Danish Staphylococcus argenteus reveals a genetically diverse collection with clear separation from Staphylococcus aureus Front Microbiol 20178151210.3389/fmicb.2017.0151228848522 PMC 5552656 · doi ↗ · pubmed ↗
- 4Witteveen S Hendrickx APA de Haan A Notermans DW Landman F et al Genetic characteristics of methicillin-resistant Staphylococcus argenteus isolates collected in the Dutch national MRSA surveillance from 2008 to 2021 Microbiol Spectr 202210 e 010352210.1128/spectrum.01035-2236005448 PMC 9603934 · doi ↗ · pubmed ↗
- 5Goswami C Fox S Holden M Leanord A Evans TJ Genomic analysis of global Staphylococcus argenteus strains reveals distinct lineages with differing virulence and antibiotic resistance gene content Front Microbiol 20211279517310.3389/fmicb.2021.79517334925305 PMC 8677677 · doi ↗ · pubmed ↗
- 6Meijer EFJ van Renssen A Maat I van der Graaf-van Bloois L Duim B et al Canine Staphylococcus argenteus: case report from the Netherlands Pathogens 20221115310.3390/pathogens 1102015335215097 PMC 8876332 · doi ↗ · pubmed ↗
- 7Wakabayashi Y Takemoto K Iwasaki S Yajima T Kido A et al Isolation and characterization of Staphylococcus argenteus strains from retail foods and slaughterhouses in Japan Int J Food Microbiol 202236310950310.1016/j.ijfoodmicro.2021.10950334968888 · doi ↗ · pubmed ↗
- 8Holt DC Holden MTG Tong SYC Castillo-Ramirez S Clarke L et al A very early-branching Staphylococcus aureus lineage lacking the carotenoid pigment staphyloxanthin Genome Biol Evol 2011388189510.1093/gbe/evr 07821813488 PMC 3175761 · doi ↗ · pubmed ↗