Differential susceptibility to colonic ulceration in mice with genetic deletion of Prostaglandin E synthase
Masako Nakanishi, Michael J. Martinez, Patrycja Sztachelski, Marion Leclerc, Daniel W. Rosenberg

TL;DR
This study shows that mice lacking a key enzyme for PGE2 production develop colonic ulcers, revealing how genetic and environmental factors influence gut inflammation.
Contribution
A novel genetic model for studying NSAID-induced colitis and interindividual variability in intestinal disease susceptibility.
Findings
Ptges-deficient mice on strain A develop spontaneous colonic ulcers, while those on C57BL/6 do not.
A/D:KO mice show increased pro-inflammatory gene expression before tissue damage occurs.
F1 hybrids of A/D:KO and B6D:KO mice are mostly free of colonic ulceration, suggesting genetic background plays a key role.
Abstract
Prostaglandin E2 (PGE2) exerts pleiotropic and context-dependent effects on inflammation, cancer and maintenance of intestinal mucosal homeostasis. To further define its role in intestinal diseases, we genetically inactivated its rate-limiting synthesis, Ptges, in two mouse lines. An unexpected phenotype consisting of spontaneous mucosal ulceration was found exclusively in the colons of strain A mice. This study aims to characterize the phenotype that may have a clinical relevance to NSAID-induced enteropathies. Mucosal ulcerations were characterized in Ptges-deficient mice maintained on strain A (A/D:KO) and C57BL/6 (B6D:KO) backgrounds. RNA sequencing of colons identified inflammatory signatures in sensitive A/D:KO mice. Microbial dysbiosis was evaluated in the fecal stream using 16S rRNA sequencing. The potential role of genetic and environmental factors in the etiology of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
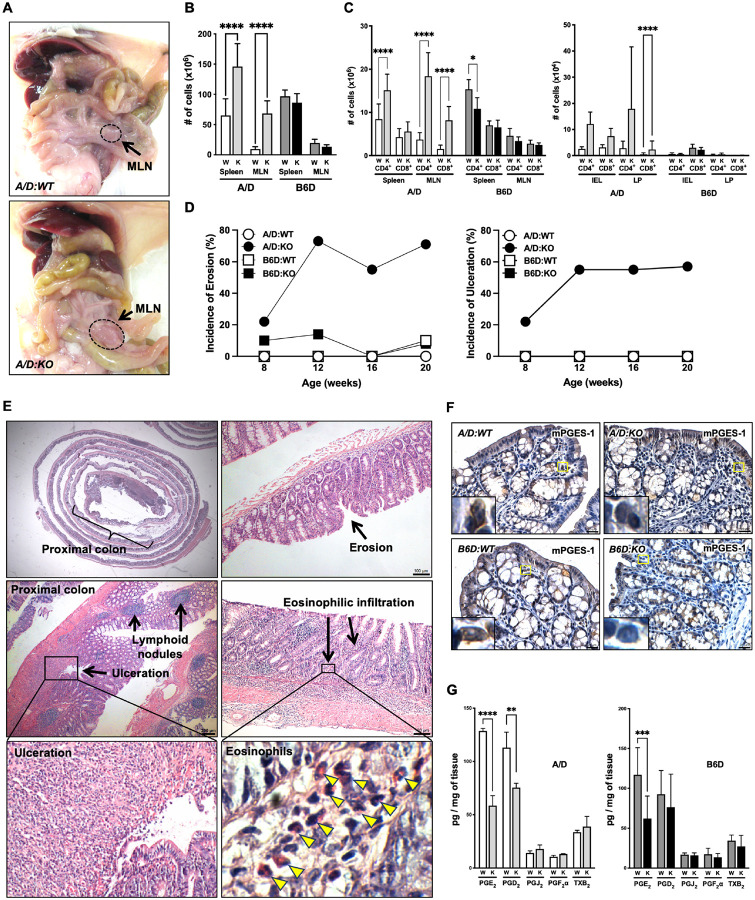 Figure 1
Figure 1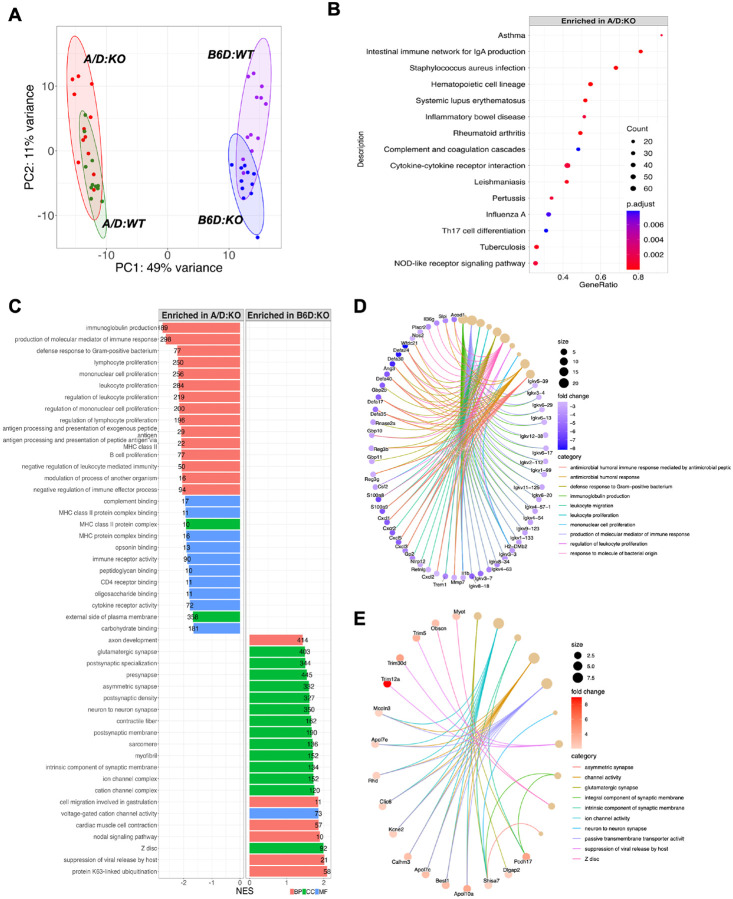 Figure 2
Figure 2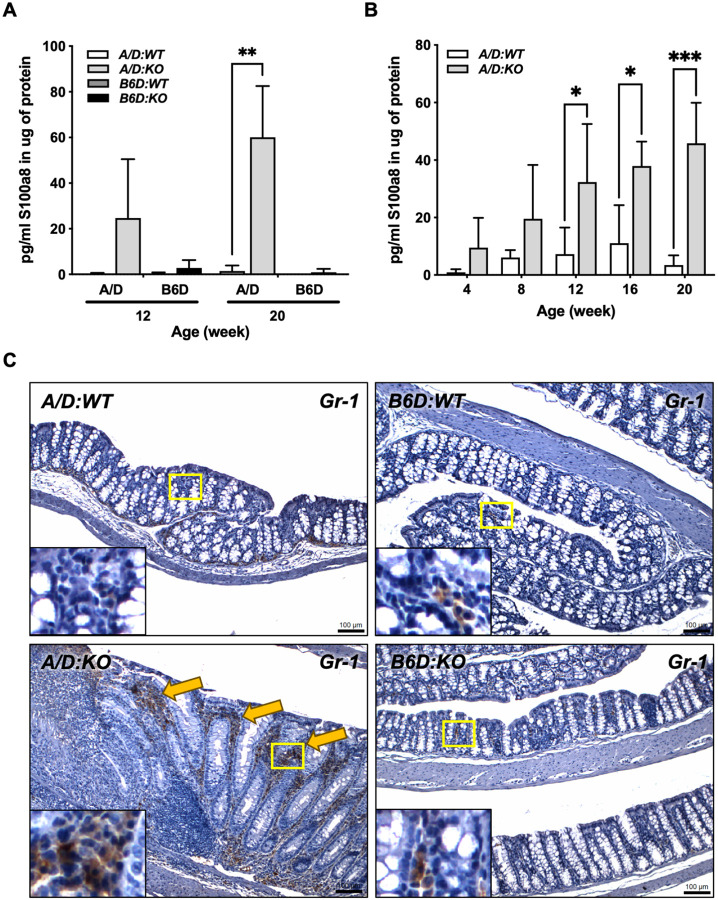 Figure 3
Figure 3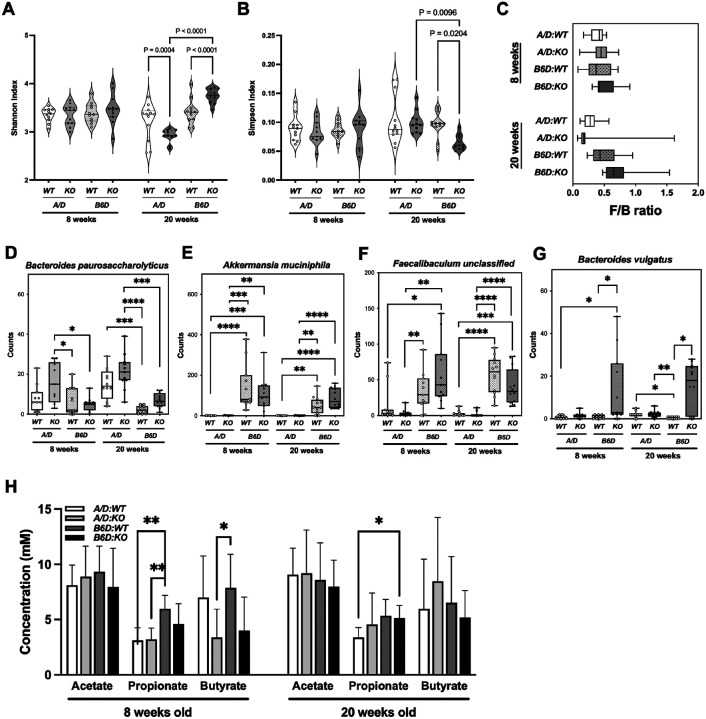 Figure 4
Figure 4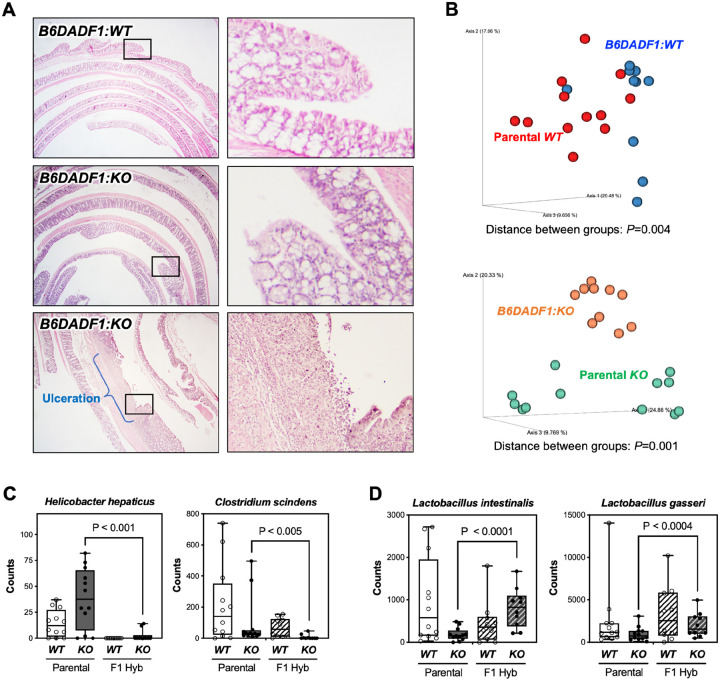 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHelicobacter pylori-related gastroenterology studies · Inflammatory mediators and NSAID effects · Inflammatory Bowel Disease
Background
The generation of the prostanoids through the arachidonic acid (AA)-cyclooxygenase (COX) metabolic pathway regulates key aspects of cellular physiology. Among these bioactive lipids, prostaglandin E_2_ (PGE_2_) is the most abundantly produced prostanoid, impacting an array of biological processes associated with inflammation, cancer and paradoxically, cell homeostasis [1]. Blockade of COX activity by commonly used non-steroidal anti-inflammatory drugs (NSAIDs) reduces PGE_2_ levels, which has proven to be an effective strategy for managing pain and inflammation, and in some cases for preventing cancer [2, 3]. However, long-term treatment with NSAIDs can cause off-target effects, including cardiovascular toxicity and gastrointestinal (GI) ulcerations [2]. In fact, a record-based retrospective single-center study showed that approximately 50% of patients taking NSAIDs reported GI-related side effects [4].
These adverse effects are due, in part, to dysregulated synthesis of related prostanoids sharing a common metabolic precursor [5]. To specifically reduce PGE_2_ levels while sparing these other essential metabolites, we [6, 7] and others [8] have targeted the inducible terminal enzyme, microsomal PGE synthase-1 (mPGES-1; Ptges), as an alternative therapy for managing inflammation and cancer.
In earlier studies, we demonstrated that genetic inactivation of Ptges provides protection against intestinal tumor development in carcinogen-treated C57BL/6 and strain A mice, affording comparable protection to targeting COX activity with NSAIDs [6, 7, 9]. During the course of these studies, however, we unexpectedly observed spontaneous ulcerations appearing throughout the colons of strain A mice lacking Ptges, a phenotype that was largely absent in C57BL/6 mice with this null genotype. This observation indicates a strain-dependent differential sensitivity to Ptges blockade in the colon that may provide a model for interindividual patient sensitivity to long-term NSAID treatment.
Use of slow-release or enteric-coated NSAIDs (e.g., Ibuprofen, naproxen), selective Cox inhibitors (e.g., Celebrex), as well as concurrent administration of proton pump inhibitors (PPI) have reduced the incidence of upper GI complications [10]. However, they fail to protect the lower GI, particularly the colonic mucosa [11]. In fact, NSAID-associated injuries to the colonic mucosa, collectively termed colopathy, display erosions, ulcerations and diaphragm-like strictures, as well as perforation, obstruction and diverticulitis [12–15]. While the overall prevalence and incidence of colopathy is unclear, approximately 10% of colitis cases are related to NSAID intake [16]. Pathogenesis of NSAID-induced colopathy remains understudied, partly due to the varying risk of developing clinically significant cases within the general population [17].
To fully characterize this highly penetrant phenotype in mice and its potential translational relevance, the following study was undertaken. We report here that the localized colonic inflammation that develops in susceptible *Ptges-*deficient strain A mice closely resembles the pathology of human drug-induced colitis. Gene expression profiling of normal colonic mucosa prior to the onset of active disease has identified dysregulation of several key signaling pathways that may contribute to mucosal injury, including the activation of inflammatory networks and antibacterial responses. While fecal microbiome analysis showed marked differences in the abundance of resident microbial taxa in the parental mouse lines, co-housing of these mice did not rescue the inflammatory phenotype in strain A, nor confer sensitivity to the colons of C57BL/6 mice. However, an F1 hybrid of these mouse lines were mostly free of colonic ulceration, indicating that genetic background may be the dominant factor in the development of the colitis-like phenotype.
Materials and Methods
Animals.
Strain A mice (A/J) and C57BL/6J (B6) were purchased from The Jackson Laboratory (Bar Harbor, ME). Ptges heterozygous (HET) mice on a C57BL/6NTac background were provided by Merck Frosst Canada, Ltd. [18] and crossed to wild-type (WT) C57BL/6J for 10 generations (B6.129P2(B6)-Ptges^tm1Dwr^/Drmn, B6D:HET), and maintained as Ptges knockout (B6D:KO) mice at the University of Connecticut Health (UConn Health). B6D:KO mice were then backcrossed to A/J mice for 10 generations at UConn Health (A. 129P2(B6)-Ptges^tm1Dwr^/Drmn, A/D:HET), and A/D Ptges knockout (A/D:KO) mice were generated by crossing A/D:HET N10 animals [6]. For co-housing experiments, two to three age-matched A/D and B6D mice were placed in a cage at 4 weeks of age and maintained for 16 weeks. Each cage housed at least 2 mice of different mouse strains and Ptges genotype for both males and females. For F1 hybrid mice, male A/D and female B6D mice were crossed once for each genotype, generating B6DADF1:WT and B6DADF1:KO mice. F1 hybrid mice were housed separately by the Ptges genotype and maintained for up to 30 weeks of age. Parental mouse lines were sacrificed at 8, 12, 16 or 20 weeks of age, and feces, whole blood, spleens, mesenteric lymph nodes (MLN) and colon tissues were collected for further analysis. A/D:WT and A/D:KO mice were littermates of A/D:HET breeding, and B6D:WT and B6D:KO mice were maintained by respective homozygous breeding. A/D and B6D mice were housed separately, except for co-housing experiments, and both male and female mice were used in the study. Only male mice were used for flow cytometry, lipidomics, gene expression analyses and S100a8 ELISA. Mice had access to maintenance diet (Teklad Global 19% Protein Extruded Rodent Diet) and drinking water ad libitum. All animal experiments were conducted with approval from the Center for Comparative Medicine (CCM) at UConn Health (AP-200208–0823).
Flow cytometry analysis.
The spleen and mesenteric lymph node (MLN) were crushed through a 100μm nylon mesh cell strainer and washed with buffered salt solution (BSS) (n = 4 per group, male mice). Splenocytes were treated with ammonium chloride to lyse red blood cells. Immune cells were extracted according to the method by Qiu and Sheridan [19] with modifications. Briefly, colons were flushed and treated with dithioerythritol (DTE) solution to extract the IEL. After removing epithelial cells with 0.5M EDTA solution, the LP was extracted by incubating the tissues with collagenase solution and Percoll gradient. Cells were fixed with 1% formaldehyde solution and stained with CD4-PerCP and CD8-PE-Cy7 antibodies (BD Biosciences). Dead cells were excluded by staining with LIVE/DEAD Cell Imaging Kit (Thermo Fisher). FACS analyses were performed on a Becton-Dickinson LSR II (Becton-Dickinson).
Histopathological evaluation.
For histopathologic analysis, colons were flushed with ice-cold PBS and opened flat longitudinally, fixed in 10% neutral buffered formalin solution overnight and stored in 70% ethanol thereafter. Formalin fixed colons were Swiss-rolled and embedded in paraffin for sectioning at 5μm thickness. Tissue sections were deparaffinized and stained with hematoxylin and eosin (H&E). The colon was evaluated across three distinct regions: proximal, middle, and distal. The proximal section of the colon is defined as the upper region to the point where the transverse colonic folds disappear. The rest of the colon was divided into two areas, with the proximal section defined as the middle colon and the remaining as the distal colon [20]. The number of erosions and ulcers were quantified for each section. Erosions were defined as colonic epithelium with reactive changes and/or localized dropout often associated with the infiltration of inflammatory cells into the submucosa without remodeling of the muscularis mucosa [21]. Ulcers were defined as loss of colonic epithelium and lamina propria was always associated with inflammatory cell infiltration of the submucosa with remodeling of the muscularis mucosa. Eosinophilic foci were defined as clusters of 25 or more eosinophils. Lymphoid lesions are composed of lymphoid follicles and colonic patches, the former corresponding to intramucosal collections of lymphocytes and the latter to collections of lymphocytes that reach deeper than the mucosal muscle plate. Within in the proximal region, fold enlargement was defined by accompanying inflammatory changes within the mucosa and submucosal tissue, and fold atrophy was defined as inconspicuous folds. Diameter of the lesions was measured from still images captured by Q-Capture Pro software. The percent of affected areas were calculated from the total diameter of the lesions divided by the entire length of the colon.
RNA sequencing.
Colons were cut longitudinally, and proximal colons were snap-frozen in liquid nitrogen and stored at −80°C for RNAseq analysis (n = 12 per group, male mice). Tissues were homogenized by Polytron Homogenizer and total RNA was extracted from the tissues using RNeasy Midi Kit (Qiagen) according to the manufacturer's instructions. Quality of RNA was assessed by 2100 Bioanalyzer system (Agilent Technologies). RNA sequencing was performed at the Center for Genome Innovation (CGI), Institute for Systems Genomics, University of Connecticut as previously described with modifications [22]. Briefly, mRNA-seq libraries were prepared using the TruSeq Standard mRNA Library Prep kit (Illumina) and sequenced on the Illumina NovaSeq 600 platform. Raw reads were trimmed using Trimmomatic/0.36. The resulting reads will be mapped to the Ensembl Mus musculus genome (GRCm38.102.gtf) using star/ 2.5.3a alignment application. Reads were quantified using featureCounts (subread/2.0.3).
Lipidomics analysis.
Colons were cut longitudinally and snap-frozen in a Precellys tube (Cayman Chemicals) in liquid nitrogen and stored at −80°C for further processing (n = 3 per group, male mice). Lipidomic analysis was performed at the Lipidomics Core at Wayne State University, as previously described [23]. Briefly, samples were homogenized and spiked with a mixture of internal standards for each individual eicosanoid (5-ng each), and biochemicals were eluted through conditioned C18 cartridges for LC-MS analysis. HPLC was performed on a Prominence XR system (Shimadzu) using a Luna C18 (3μ, 2.1×150 mm) column. HPLC eluate was directly introduced into the electrospray ionization source of a QTRAP5500 mass analyzer (SCIEX) in the negative ion mode with Multiple Reaction Monitoring (MRM). Data were collected and quantitated using Analyst 1.6.2 (SCIEX) and MultiQuant (SCIEX) software, respectively. Correction for recovery efficiencies and relative quantitation of each analyte were performed using signals in each chromatogram corresponding to the spiked-in internal standards.
Microbiome analysis.
Fecal samples from 8- and 20-week-old mice were collected and stored at −80°C (n = 6–10 per group). Microbial DNA was extracted from fecal samples using the Shoreline Complete kit (Shoreline Biome, Farmington, CT) or Fast DNA stool mini kit (Qiagen), according to the manufacturer's protocols. For the parental mouse lines, StrainID amplicons (~ 2500 base pairs) were sequenced that encompass the entire 16S-rRNA gene, the highly variable internally transcribed spacer (ITS) region, and a portion of the 23S-rRNA gene to increase taxonomic resolution. Primers used for the amplicon synthesis had a 3-part sequence (5'-adaptor–barcode–target-specific primer-3') as described previously [24]. Briefly, the 16S adaptor sequence is 5'-GGTTATGCGGTTCACTGC-3', all barcodes were selected from the list of 384 Pacific Biosciences (PacBio)-recommended barcodes (https://www.pacb.com/products-and-services/analytical-software/multiplexing/). The target-specific forward primer sequences are a pool of primers with the sequence 5'-AGRRTTYGATYHTDGYTYAG-3'. The reverse primer had a similar 5'-adaptor–barcode–target-specific primer-3' structure, where the adaptor sequence is 5'-CGTCACTTGGCGTATTGG-3', and the target-specific sequences are a pool with the sequence 5'-AGTACYRHRARGGAANGR-3'. SMRTbell adaptors were added to pooled samples and sequencing was conducted on a PacBio Sequel 1 system. Raw sequence reads were demultiplexed using SBAnalyzer software and assigned taxonomy with strain-level resolution using the Athena Microbial Reference Database (Shoreline Biome).
Microbiome analysis of additional parental lines, co-housing and F1 hybrid mice were performed by 16S rRNA sequencing at Microbiome Research Core, University of Alabama at Birmingham.
Short chain fatty acid (SCFA) analysis.
Fecal samples (≥ 20 mg) were collected from individual mice and stored at −80°C (n = 10 per group). Analysis was performed at the Host-Microbe Metabolomics Facility (HMMF) at The University of Chicago, as previously described [25]. Briefly, short chain fatty acids were derivatized with pentafluorobenzyl-bromide (PFBBr) and analyzed via gas chromatography-mass spectrometry (GC-MS, Agilent 8890) with chemical ionization (CI) with negative mode detection. The oven ramp parameters was 1 min hold at 60 ÅãC, 25 ÅãC/min up to 300 ÅãC with a 2.5 min hold at 300 ÅãC. Inlet temperature was 280 ÅãC and transfer line was 310 ÅãC. Using a HP-5ms ultra inert column (30 m × 0.25mm, 0.25 μm; Agilent Technologies 19091S-433UI), methane as the reagent gas. The instrument was in Scan acquisition mode (m/z 50–600) with a solvent delay of 4.2 with Normal Scanning and operating in negative chemical ionization mode with methane used as the reagent gas (99.999% pure). For data transformation, a 10-point calibration curve was prepared with acetate (100 mM), propionate (25 mM), and butyrate (12.5 mM) with 9 subsequent 2x serial dilutions. Data analysis was performed using MassHunter Quantitative Analysis software (version B.10, Agilent Technologies) and confirmed by comparison to retention time, nominal mass, and fragmentation patterns of authentic standards. Normalized peak areas were calculated by dividing raw peak areas of targeted analytes by raw peak areas of spiked internal standards (Cambridge Isotope Laboratories, D3-acetate DLM-3126–25; D5-propionate DLM-1601–1; D7-butyrate DLM-7616-PK; D6-phenol DLM-370-PK). Relative abundance data were normalized to the average of D7-proline/D6-phenol. Authentic standards were purchased for all targeted compounds and prepared at 1 mg/ml in methanol. All metabolite identifications are level 1 according to the CAWG standard initiative [26].
Immunohistochemistry.
Tissue sections were deparaffinized and subjected to heat-induced antigen retrieval in sodium citrate buffer. Tissues were then incubated overnight with primary antibody for mPGES-1 (1:4000, PAB0130, Abnova) or Gr-1 (1:4000, RB6–8C5, Invitrogen), followed by an incubation with HRP-conjugated secondary antibody (Cell Signaling), and the signal was detected with DAB (Vector Laboratories) and counter-stained with hematoxylin. Images were captured using a conventional microscope using Q-capture Pro software.
ELISA assays.
For S100a8 (Calprotectin) analysis, serum was collected in serum separator additive microcentrifuge tubes (BD) and centrifuged at 15,000 RCF for 90 seconds and stored at −80°C. Fecal samples were collected and stored at −80°C (n = 3–4 per group, male mice). Total protein from feces was extracted in a protein extraction buffer (50mM Tris, pH 7.5, 150mM NaCl) and filtered through 0.2μM column (Costar). S100a8 levels in serum and fecal samples were measured using Mouse S100A8 DuoSet ELISA (R&D Systems) according to the manufacturer's protocol.
Statistical analysis.
Statistical analyses were performed using GraphPad Prism 10 software (GraphPad Software, Inc., La Jolla, CA) for FACS, histopathology, lipidomics, ELISA and SCFA analyses. Data are presented as the means ± SEM, and analyses were performed by one-way ANOVA with a Bonferroni comparison test. A P-value less than .05 (P < .05) was considered statistically significant. For RNAseq analyses, differentially expressed gene analysis was performed using DESeq2 R-package (version 1.42.0) with the reference level (denominator) set to A/D:KO. Adjusted P-values of the tests (adj. P < .05) were used to determine the significance for all analyses. GSEA were performed using gseGO and gseKEGG in ClusterProfiler, v4.9.0.002[27] with Benjamini-Hochberg corrected P < 0.05 as the inclusion cutoff. For microbiome analyses of parental lines, a series of custom scripts were used to survey the bacterial community compositions at the phylum, family, and species levels. Sample alpha diversity was assessed using Shannon's and Simpson's indices. Pairwise comparisons of alpha diversity were conducted using ANOVA with Tukey HSD for post-hoc testing. Differential abundance analysis was conducted at the family and species level using R package MaAsLin2[28] with “group” (A/D and B6D) as a fixed effect. For additional parental lines, co-housing and F1 hybrid mice, analysis of alpha (Shannon) and beta (Bray-Curtis) diversities were performed using R packages phyloseq as described previously [29]. Pairwise PERMANOVA and/or Kruskal-Wallis test was used to evaluate between-group differences. A P-value less than 0.05 (P<0.05) was considered statistically significant.
Results
A/D:KO mice develop spontaneous colonic ulcerations
We reported previously that global deficiency of Ptges in strain A mice (A/D:KO) caused localized mucosal ulcerations affecting up to 15% of the colonic epithelium, while there were no obvious phenotypic defects observed in C57BL/6 mice with the same genetic deletion (B6D:KO) [6, 7]. To characterize the distinct phenotype in detail, we first performed gross examination and physical evaluation from 8 to 20 weeks of age. As shown in Fig. 1A, A/D:KO mice had markedly enlarged mesenteric lymph nodes (MLN) compared to the A/D:WT mice at 20 weeks of age. Swelling of the MLN appeared as early as 8 weeks of age in the A/D:KO mice and was found more frequently over time (data not shown).
To assess the extent of inflammation, spleens and MLN, colonic intraepithelial lymphocytes (IEL), and lamina propria (LP) were extracted from 20-week-old mice and examined for the presence of CD4^+^ and CD8^+^ T cells. As shown in Fig. 1B, A/D:KO mice had significantly more T cells within the MLN compared to WT littermates. In addition, A/D:KO mice had the most significantly elevated levels of CD4^+^ and CD8^+^ cells within the MLN (Fig. 1C). In the colonic mucosa, there was a trend towards increased levels of these immune cells, with a significant increase observed in the levels of CD8^+^ cells within the LP compartment (Fig. 1C). There was no concomitant changes observed in these markers in the B6D:KO compared to WT counterparts.
To further characterize the histological expansion over time, colons were examined at 8, 12, 16 and 20 weeks of age. Colonic lesions were characterized by the extent of epithelial cell erosion and overall ulceration as described in Materials and Methods. While B6D:KO mice showed only minimal mucosal erosion, A/D:KO mice developed multiple colonic lesions at 8 weeks, progressing over time (Fig. 1D). Ulcerations were mostly confined to the proximal colon, although occasionally extending throughout the colon with associated lymphoid nodules and patches of mucosal erosion (Fig. 1E). Moreover, intraductal glandular abscesses/erosions were infiltrated with scattered eosinophils, associated with early stages of mucosal damage (Fig. 1E, arrowheads). However, the lesions were typically minimal-moderate, affecting less than 10% of the entire colon. This observation may explain why these mice rarely show overt symptoms of colitis, including rectal breeding and prolapse, even at 20 weeks of age.
We next examined the impact of Ptges deficiency on the local formation of PGE_2_ and the possibility of metabolite redirection towards other prostanoids [30]. A panel of prostanoids were measured in the colons of these mice at 10 weeks of age, prior to the appearance of detectable colonic ulcerations. Both strains of KO mice showed a complete loss of mPGES-1 protein expression (Fig. 1F) [6, 31], and the tissue levels of PGE_2_ were reduced to a comparable extent between these mice. There was only minimal metabolic shunting towards PGJ_2_, PGF_2_ or TXB_2_, although PGD_2_ was reduced significantly in the A/D:KO mice (Fig. 1G). Levels of PGD_2_ varied in Ptges deficient mice as increased levels of PGD_2_ have been reported in lung and macrophages [30], as well as in the stomach [32]. Functionally, PGD_2_ has been shown to have anti-inflammatory properties [33]; however, it also acts as a potent eosinophil chemoattractant during allergic reactions, thereby exhibiting pro-inflammatory properties [34]. In fact, A/D:KO mice show an influx of eosinophils in the colon as early as 8 weeks of age. The potential effects of PGD_2_ and eosinophils on the development of inflammatory phenotype in the A/D:KO warrant further investigation.
An early-onset inflammatory response in the A/D:KO may drive the development of colonic ulceration
To identify differentially expressed genes (DEGs) associated with the divergent disease susceptibility in the two mouse lines, we performed bulk RNA sequencing (RNAseq) analysis on proximal colons of 10-week-old A/D and B6D mice, with or without Ptges. This is the time point immediately prior to the appearance of visible mucosal damage, in which 2 out of 9 mice developed even mild ulcerations covering less than 1% in their colonic mucosa.
While intrastrain comparisons identified fewer changes between WT and KO for A/D vs. B6D (46 genes vs. 255 genes respectively; Table 1), interstrain comparisons revealed more impressive DEGs regardless of Ptges genotype (A/D:WT vs. B6D:WT, 1262 genes; A/D:KO vs. B6D:KO, 942 genes; Table 1), evident in PCA plots that showed complete separation between the two mouse lines (Fig. 2A). Despite fewer DEGs, Ptges status produced a wider spread of data points in the A/D compared to B6D mice (Fig. 2A), suggesting that genetic deletion of Ptges causes highly variable but modest levels of gene expression changes in the A/D line compared to B6D mice.
Comparing between A/D:KO vs. B6D:KO mice, KEGG pathway analysis revealed that the DEGs enriched in A/D:KO colons were mainly involved in inflammatory pathways, including asthma, intestinal immune network for IgA production and inflammatory bowel disease (Fig. 2B). These pathways affect various cellular processes, including mast cell activation, neutrophil/macrophage chemotaxis and modulation of antimicrobial peptides. Furthermore, Gene Ontology (GO) analysis showed that inactivation of Ptges in the A/D colon was predominantly associated with biological processes centered around the immune response against bacteria and/or mediated by antimicrobial peptides (Fig. 2C). In addition, these functional changes were accompanied by molecular alterations to key immune-related responses, including cytokine/chemokine activity and complement binding (Fig. 2C). B6D:KO colons also showed the enrichment of several functional activities related to immune response, although these pathway alterations were distinct from those present in A/D:KO mice. For example, pathways enhanced in the B6D:KO mice included suppression of viral release, regulation of lipid digestion, cholesterol absorption and the general process of post-translational modification (e.g., K63-linked ubiquitination) (Fig. 2C). Notably, several genes were associated with membrane trafficking activities involving transporters and ion channels (Fig. 2C). Cellular interactions and receptor signaling pathways support many essential functions during the inflammatory response. Thus, it is possible that some of these gene signatures represent the activation of molecular pathways in the B6:KO mice that contribute to the maintenance of mucosal homeostasis despite reduced PGE_2_ levels.
To gain further insight into underlying DEGs comprising these pathways, a Gene-Concept Network plot (Cnet) was generated using the top 10 GO pathways. As shown in Fig. 2D, one of the striking signatures in A/D:KO mice was a wide range of genes encoding the immunoglobulin (Ig) heavy and light chains, perhaps indicating the rapid activation of plasma cells associated with an adaptive immune response. This robust expression of Ig repertoires may be the basis for the extensive variability profile in PCA plot of the A/D:KO mice (Fig. 2A).
As shown in Fig. 2D, some of the most highly enriched genes within the A/D:KO colons included pro-inflammatory markers (S100a8 and S100a9) and chemokines (Cxcl2, Cxcl3, Cxcl5), as well as various Ig genes. S100a8 and S100a9 are well-characterized pro-inflammatory proteins that are a hallmark of inflammatory diseases [35], collectively referred to as the calprotectins [36]. Fecal calprotectin levels have been shown to correlate with clinical disease activity in inflammatory bowel disease (IBD), and as such have been widely used as a biomarker for the disease [37]. Playing a crucial role in the inflammatory response, the S100a8/S100a9 complex demonstrates potent antimicrobial properties [38]. Similarly, Cxcl2, Cxcl3 and Cxcl5 are not only potent chemoattractants for neutrophil migration [39], but also display antimicrobial activity as well [40]. Indeed, the A/D:KO colons were also enriched with several prominent antimicrobial peptides (AMPs), including α-defensins (Defa24, Defa30, Defa40, Defa17, Defa35) and regenerating islet-derived protein 3-y (Reg3y) (Fig. 2D). α-defensins, also referred to in the mouse as the cryptdins, are proteolytically activated by matrix metalloproteinase 7 (Mmp-7) [41], which was also increased in the A/D:KO colons (Fig. 2D). Interestingly, Mmp-7 deficient mice show a significant decrease in PC-derived α-defensin expression, which in turn causes dysbiosis [42]. The exaggerated antibacterial response instigated by Ptges blockade in A/D mice suggests that their mucosal homeostasis is impaired upon their early interactions with gut bacteria.
In the colons of B6D:KO mice, many genes were associated with protection against tissue injury, including tripartite motif genes (Trim12a, Trim30d and Trim5) (Fig. 2E). While TRIM family of proteins regulates mucosal barrier function, they also display antiviral and antibacterial properties, directly linked to the IBD pathogenesis [43]. Moreover, expression of several Apolipoprotein L (Apol7e, Apol7c and Apol10a) genes was significantly upregulated in the B6:KO mice (Fig. 2E). Apol proteins serve as ion channels of intracellular membranes, which contribute to apoptosis in myeloid lineage and endothelial cells [44]. It has been reported that Apol proteins induced via Toll-like receptors (TLRs) promoted apoptosis in dendritic cells (DCs) to prevent overactivation of the immune system [45, 46].
Fecal S100a8 correlates with the influx of neutrophils into the colons of A/D:KO mice
To examine potential changes in the inflammatory signature over time, we measured the levels of S100a8 in the serum and in fecal samples collected at varying times relative to the onset of colonic ulceration. While S100a8 was not detected in the serum (data not shown), A/D:KO mice had significantly elevated levels of fecal S100a8 at 12 and 20 weeks of age compared to the WT mice, whereas B6D mice of either Ptges genotype showed only minimal levels (Fig. 3A). Moreover, the levels of fecal S100a8 in A/D:KO mice increased over time (Fig. 3B), clearly correlated with disease activity (Fig. 1D). In addition, colon tissues from 20-week-old mice were examined for the presence of Gr-1^+^ neutrophils by IHC. As shown in Fig. 3C, an influx of Gr-1^+^ cells were found within the ulcerated areas of the A/D:KO colons, while only few positive cells were present in the colons of other groups.
These results suggest that an inflammatory response is initiated early in A/D:KO mice, and the resistant B6D:KO mice had vastly different pathway profiles from the A/D:KO mice, indicating that these two mouse lines activate distinct signaling systems to cope with the impaired PGE_2_ synthesis. Importantly, pathway analyses have clearly indicated the involvement of the gut microbiome in the development of colonic ulcerations observed in the A/D mice.
Microbial community structure is affected by genetic background and Ptges genotype
Expression of genes involved in antibacterial response and barrier functions in the A/D:KO and B6D:KO colons suggest that the ensuing inflammatory phenotype may be correlated with a pathogenic microbial community structure. To explore the potential influence of the gut microbiome on the development of colonic ulcerations in the A/D:KO mice, we examined fecal samples collected from young adult (8-week) and mature adult (20-week) mice by 16S rRNA sequencing, as described under Materials and Methods.
As shown in Fig. 4A, alpha diversity measured by the Shannon index indicated a significant enrichment in overall microbial richness at species levels in the B6D:KO compared to the A/D:KO mice at 20 weeks (P < 0.0001). In addition, while A/D:KO mice showed a reduction, B6D:KO mice increased in microbial richness compared to their respective WT counterpart (Fig. 4A). Furthermore, the Simpson diversity index revealed that at 20 weeks, B6D:KO mice showed a significantly more enriched bacterial community at this level compared to A/D:KO mice (Fig. 4B; P = 0.0096). These results demonstrate that by 20 weeks, Ptges status, host genetic background and the presence of gut inflammation combine to exert a marked influence over the expansion/suppression of distinct bacterial species.
The ratio between the two dominant bacterial phyla (Firmicutes/Bacteroidetes, F/B ratio) are often used as a biomarker of intestinal homeostasis [47]. At 20 weeks, A/D mice exhibited a reduced F/B ratio reminiscent of IBD [47], further trending downward with Ptges deletion (Fig. 4C).
Several bacterial species within Phylum Bacteroidetes showed pronounced increase in the A/D mice including Bacteroides paurosaccharolyticus (Fig. 4D) and Rufibacter unclassified spp. Consistent with greater bacterial diversity (Fig. 4A), a wider array of bacterial species was present in B6D mice. For example, Akkermansia municiphila (Fig. 4E), was dramatically reduced within the A/D fecal stream. Akkermansia muciniphila degrades mucin for its source of carbon, nitrogen, and energy for survival [48], and serve as a major producer of short-chain fatty acids (SCFAs), including acetate and propionate, which in turn supports the growth of additional bacterial strains that produce butyrate [49]. SCFAs play a critical role in regulating gut homeostasis, and intestinal bacteria are the main source of these organic acids via degradation of dietary fibers and resistant starches [50]. In fact, several other SCFA-producing bacteria displayed distinct signatures between the four groups. For example, A/D mice harbored significantly less Faecalibaculum spp. (Fig. 4F), an established butyrate producer [51], which has been shown to reduce inflammation, in part by stimulating colonic regulatory T cells [51]. Furthermore, Bacteroides vulgatus, showed a significant expansion in the B6D:KO mice (Fig. 4G). Interestingly, Bacteroides vulgatus has been reported to attenuate the severity of DSS-induced colitis by inhibiting pro-inflammatory cytokines and modulating the growth of other SCFA-producing bacteria [52].
To determine whether these distinct bacterial signatures might be associated with the levels of SCFAs, fecal samples were analyzed by GC-MS as described in Materials and Methods. As shown in Fig. 4H, A/D:WT and A/D:KO mice had significantly lower levels of propionate at 8 weeks of age compared to B6D:WT mice. In addition, Ptges-deficient mice showed a trend towards lower butyrate levels in younger mice, an effect that disappeared as the mice aged (Fig. 4H).
Taken together, these results suggest that the A/D and B6D mice primarily harbor distinct bacterial populations, with further modifications by the absence of Ptges. Resultant bacterial profiles in turn are correlated with the early production of SCFAs, which may affect mucosal homeostasis later in life.
Co-housing of A/D and B6D mice does not alter the inflammatory phenotype
Based on the distinct bacterial signatures present in these two parental mouse lines, the following cohousing study was undertaken to determine whether the microbiome present in resistant B6D mice might harbor protective bacteria that could be transferred to the sensitive A/D mice. The other possibility is that transfer of the A/D microbiome may sensitize B6D mice to mucosal damage. It has previously been shown in C57BL/6 mice obtained from different vendors that co-housing results in a nearly complete normalization of the microbiome within 4 weeks [53]. Thus, we co-housed 4-week old A/D and B6D mice of each Ptges genotype for a total of 16 weeks (A/D:WT, n = 6; A/D:KO, n = 10; B6D:WT, n = 8; B6D:KO, n = 8). Fecal samples were collected before (4-weeks old) and after (20-weeks old) co-housing. As shown in Table 2, fecal microbial analysis revealed a significant decrease in overall bacterial richness (P = 0.038, Shannon index) in A/D:KO mice, but a significant expansion in the abundance of microbes (Bray-Curtis dissimilarity index) in the A/D mice, with (P = 0.018) and without (P = 0.001) Ptges, suggesting that bacterial transfer occurred between the co-housed mice. Indeed, we identified several bacteria using Kruskal-Wallis test that were significantly decreased in A/D:KO mice after co-housing have been reported to exacerbate inflammatory conditions such as IBD, including Bacteroides massiliensis [53], Lachnoclostridium [54], and Desulfovibrio [55]. However, others have shown bidirectional effects on inflammation such as Prevotella copri [56]. Interestingly, there was an acquired presence of a number of bacteria in A/D:KO mice after co-housing, including Akkermansia muciniphila and Bacteroides vulgatus (Table 3).
Despite the successful horizontal transfer of microbiota between the two mouse lines, however, altered microbial community structure were likely insufficient to override the overall strain-related mucosal inflammation. At 20 weeks of age, 85% (11 out of 13) of the A/D:KO mice of either sex developed colonic ulcerations after co-housing with B6D line that mirrored the parental lines.
F1 hybrid mice are protected from developing ulceration
We next asked whether mouse genetic background may affect the differential susceptibility to colonic ulceration. To determine the effects of gene dosage, we generated F1 hybrids by crossing A/D and B6D mice, with or without Ptges (B6DADF1:WT, n = 29 and B6DADF1:KO, n = 30). To standardize the effects of the gut microbiome in this study, we used female B6D mice for breeding each genotype, as the gut microbiome is primarily established through maternal transmission [57]. At 20 weeks of age, B6DADF1:WT and B6DADF1:KO mice were examined for the presence of colonic ulcerations. None of these mice exhibited enlarged spleen or MLN. Histological evaluation of the colons from B6DADF1:WT mice revealed no evidence of ulceration (Fig. 5A). A single B6DADF1:WT mouse exhibited a minute focus of eosinophils localized between the muscle layers, with a frequency likely within the range of a non-pathological condition [58]. In the B6DADF1:KO mice, one mouse showed a massive immune response with focal ulceration, and another presented with small lesion of much lesser extent compared to the parental A/D:KO mice (Fig. 5A). However, the remaining B6DADF1:KO mice (93%) were entirely free of colonic ulcerations, indicating nearly complete protection (Fig. 5A). To determine whether aging may affect disease onset, a subset of F1 hybrid mice (n = 30) were maintained for up to 30 weeks of age, but mice of either Ptges genotype remained ulcer-free (data not shown). These results indicate that the effects of genetic background have significant impact on the phenotypic variation in these mice despite impaired PGE_2_ synthesis.
To further determine the role of host-microbe interaction towards protection of colonic ulceration, we analyzed fecal samples collected from the 20-week-old F1 hybrid mice (n = 10 per group). Fecal microbiome analysis was performed by comparing between the combined parental lines (A/D and B6D) and F1 hybrids for both genotypes to identify overall changes in bacterial communities. Compared to the parental lines, microbial richness (Shannon Index) was mostly normalized at every taxonomic level in both B6DADF1:WT and B6DADF1:KO. However, the Bray-Curtis dissimilarity index indicated that bacterial community structures were significantly different between the parental lines and F1 hybrids for both genotypes (Fig. 5B; Parental WT vs. B6DADF1:WT, P = 0.004 and Parental KO vs. B6DADF1:KO, P = 0.001), suggesting that F1 hybrid lines harbor distinct microbiome. To clarify the influence of changes to microbial beta diversity on protection afforded to the colonic mucosa in the F1 hybrids, we further analyzed the data using Kruskal-Wallis test to identify bacteria that were significantly modified between the combined parental KO and B6DADF1:KO mice (Table 4). Indeed, several pro-inflammatory bacteria, including Desulfovibrionaceae [59], Roseburia [60] and Helicobacter hepaticus [61] were significantly reduced in the B6DADF1:KO mice (Table 4 & Fig. 5C). However, bacteria associated with antiinflammatory functions such as Clostridium scindens [62] and Oscillibacter [63] were also decreased in the B6DADF1:KO mice (Fig. 5C). Interestingly, the majority of the bacteria that were significantly increased in the B6DADF1:KO mice have been reported to alleviate colonic inflammation, including Lactobacillus intestinalis [64], Lactobacillus gasseri [65], Muribaculaceae [66], and Dubosiella [67] (Table 4 & Fig. 5D). In addition, a recent study has found that Lactobacillus vaginalis, traditionally known for its role in reducing vaginal inflammation, also ameliorates DSS-induced colitis by regulating gut microbiota balance and restoring intestinal barrier function [68]. Overall, the shift in microbial community structure in the F1 hybrid mice favors an anti-inflammatory milieu, highlighting their potential as candidates for probiotic therapy. However, a limitation of the current study is that the fecal microbiome analysis has provided only a snapshot of overall community structure present within the gut [69]. Ultimately, the potential role of altered microbial communities in the instigation of colonic ulceration should be carefully explored in future studies to capture spatially distinct niche that may be contributing to the disease onset.
Discussion
PGE_2_ regulates discrete phases of the acute inflammatory process, including promotion of neutrophil recruitment and pro-inflammatory cytokine production as inflammation is underway [70]. Following the acute phase of tissue injury, PGE_2_ then elicits a robust immunosuppression that contributes to epithelial resolution, enabling repair and tissue regeneration [70]. Confirmation of the context-specific roles of PGE_2_ during the inflammatory response has been demonstrated in several preclinical models. For example, *Ptges-*deficient mice exhibit attenuated inflammation upon LPS challenge [18] but also display delayed recovery from tissue injury in acetic acid-treated gastric ulcers [71], as well as following dextran sulfate sodium (DSS)-induced colitis [72].
While no obvious mucosal defects have been observed with naive Ptges KO mice maintained on commonly used C57BL/6 [7, 18] or DBA/1LacJ [73] backgrounds, an introduction of the same genetic blockade to strain A mice causes spontaneous colonic ulceration, with mucosal damage occurring as early as 8-weeks of age. Such a marked interstrain response to the physiological stress induced by reduced PGE_2_ levels underscores the role of genetic variability in inbred mice that contributes to disease susceptibility in the colon. In earlier studies that evaluated the role of IL-10 deficiency on the development of enterocolitis [74], a widely divergent disease susceptibility was found that was entirely dependent upon genetic background [75]. For example, IL-10 deficiency in C3H/HeJBir and 129/SvEv mice induced severe colitis, whereas BALB/c and 129xC57BL/6J mice developed an intermediate phenotype [76]. In contrast, IL-10:KO mice on C57BL/6J background developed only mild intestinal disease with delayed onset [76]. Interestingly, strain A mice phylogenetically related to BALB/c mice [77], and thus the shared sensitivity to induced colitis, albeit driven by different genetic stressors, may provide clues to the underlying genetics that may contribute to enhanced sensitivity.
A well-documented genetic aberration that contributes to susceptibility to inflammation in strain A mice is a loss-of-function mutation in complement component 5 (C5) [78]. Lack of C5 impairs production of pro-inflammatory cytokines from macrophages, predisposing the mice to aberrant pathogenic infections [78]. Moreover, a potentially crucial difference between strain A and B6 mice relates to their fundamental differences in the underlying biology of T helper cells. For example, whereas B6 mice generally mount a potent Th1-type response to pathogenic infections, strain A mice tend to develop a Th2-predominant response [79]. Indeed, our RNAseq data (Fig. 2B) highlight increases in a set of genes involved in complement binding and Th2-triggering events, including asthma and susceptibility to bacterial infections in the A/D:KO colons, gene signatures that were absent in B6D:KO mice. While Th2 cells contribute to repair and tissue regeneration, their excessive tissue infiltration can induce pathological fibrosis [80]. These earlier observations combined with the results of the present study suggest that A/D mice may be inherently hypersensitive to otherwise inconsequential environmental insults, generating, in turn, mucosal 'microinjuries', a direct consequence of a Th2-skewed immune system. Considering the regulatory role of PGE_2_ in controlling key aspects of inflammation, genetic disruption of Ptges in strain A mice may further drive subclinical inflammatory towards overt mucosal injury. In fact, a broad panel of inflammatory chemokines (Cxcls) required for neutrophil recruitment were significantly upregulated in A/D:KO mice prior to the appearance of discernible ulcerations (Fig. 2D), accompanied by an influx of Gr-1^+^ neutrophils surrounding the ulcerated lesions (Fig. 3C). Considering a possible threshold level of PGE_2_ that is required for neutrophil clearance, our findings suggest that continuous recruitment of neutrophils into the affected mucosa can trigger fulminant tissue damage [81]. Accumulation of these microinjuries in the tissue over time may ultimately present as more extensive mucosal damage, as evident in the A/D:KO mice.
Most importantly, the F1 hybrid mice (B6DADF1:KO), despite maintaining impaired PGE_2_ synthesis, demonstrated nearly complete resistance to colonic ulceration. This finding indicates that the parental B6D line has conferred protection to the F1 mice. On the other hand, the underlying susceptibility to colitis in the A/D line was masked in the progeny. C57BL/6 mice exhibit varying degree of resistance and susceptibility to intestinal inflammation. For example, Bouma et al. [82] reported that F1 hybrid mice of colitis-resistant C57BL/6 and susceptible SJL/J developed an intermediate phenotype in response to trinitrobenzene sulfonic acid (TNBS), although further resistance was present in the F2 generation. On the other hand, in the CD40-driving colitis model, F1 mice of C57BL/6 (sensitive) and BALB/c (resistant) did not show evidence of histopathologic alterations in the colon [83]. Compared to models that induce severe colitis, the extent of colonic ulceration in the A/D:KO mice is less severe that one or more susceptibility genes, if present, could have been inherited as recessive traits.
Alternatively, we cannot exclude the possibility that gene dosage has impacted the gut microbial community structure, in turn altering the luminal metabolome, which may have contributed to gut homeostasis in the F1 mice. It is well-established that host genetics plays an important role in shaping the gut microbiome, which, in turn, is implicated in the development of IBD-related pathologies [84]. In one such study, a combine deficiency of Nod2 and Cybb, known Crohn's disease susceptibility genes, caused the accumulation of the pathobiont, Mucispirillum scheaedleri, that triggered the onset of colitis in a mouse model [85]. Moreover, Lamas et al. [86] tested another IBD susceptibility gene, Card9, that promotes recovery from mucosal injury by stimulating IL-22. Interestingly, *Card9-*deficient mice were found to be susceptible to colitis due to impaired tryptophan metabolism, mitigating the formation of aryl hydrocarbon receptor (AHR) ligands [86]. The administration of Lactobacillus strains was sufficient to reduce colitis in the Card9:KO mice [86], demonstrating the complex interaction between host genetics, microbial community structure and metabolite balance that collaborate to maintain intestinal homeostasis.
Whether gene-microbe interactions may play a role in Ptges-dependent colitis is not presently known. Indeed, the microbial composition present in the B6DADF1:KO were significantly different from that found in either parental line. Analysis of the community structure in the F1 hybrids identified increased abundance of several bacteria (Table 4) that are associated with protection against experimentally-induced intestinal inflammation. For example, colonization of Lactobacillus intestinalis has been shown to alleviate DSS-induced colitis in mice by inhibiting the production of serum amyloid A proteins (SAAs) from intestinal epithelial cells, leading to the suppression of Th17 differentiation [64]. Within the same family of bacteria, Lactobacillus gasseri is an established probiotic that contributes to the maintenance of healthy gut function [87].
Interestingly, several of the bacteria present in the parental A/D:KO feces, including Bacteroidetes and Proteobacteria, are also enriched in mice treated with various NSAIDs such as indomethacin, naproxen, and diclofenac [88]. On the other hand, Lactobacillus and Bifidobacterium families of bacteria have been tested for their probiotic efficacy to reduce NSAID-induced intestinal injuries. For example, administration of the probiotics, Lactobacillus casei and Lactobacillus paracasei, can ameliorate clinical signs of intestinal inflammation induced by indomethacin in mice [89]. Moreover, a recent double-blind, placebo-controlled trial has shown that Bifidobacterium breve reduces intestinal damage caused by an 8-week daily intake of low-dose acetylsalicylic acid (ASA) in healthy volunteers[90]. Based on these findings, several bacterial species found in the F1 hybrid mice, including Lactobacillus intestinalis, Lactobacillus gasseri and Bifidobacterium sp. (Table 4 & Fig. 5D) may be further evaluated as candidate probiotics for the treatment of NSAID-induced enteropathy.
Conclusions
In summary, we have reported the development of a strain-dependent enteropathy in strain A mice that recapitulates the histologic features of NSAID-induced injury that occurs in a subset of patients on long-term drug treatment. Our results suggest that the absence of mPGES-1 activity, with associated reduced levels of PGE_2_, causes a dramatic shift in the inflammatory milieu in A/D:KO mice. On the other hand, the resistance to mucosal injury observed in B6D:KO mice underscores their ability to maintain mucosal homeostasis despite compromised PGE_2_, which may have played a dominant role in the F1 hybrid mice. Our findings warrant further studies to define the complex networks connecting inducible PGE_2_ synthesis with gene-microbe interactions. Considering the already compromised levels of PGE_2_, these genetically modified mice may serve as a useful model to study potential mechanisms to explain why a subset of patients are at greater risk of developing mucosal ulceration upon NSAID therapy.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nakanishi M, Rosenberg DW. Multifaceted roles of PGE 2 in inflammation and cancer. Semin Immunopathol. 2013;35(2):123–37.22996682 10.1007/s 00281-012-0342-8PMC 3568185 · doi ↗ · pubmed ↗
- 2Wang D, Du Bois RN. The role of anti-inflammatory drugs in colorectal cancer. Annual review of medicine. 2013;64:131–44.10.1146/annurev-med-112211-15433023020877 · doi ↗ · pubmed ↗
- 3Drew DA, Goh G, Mo A, Grady JJ, Forouhar F, Egan G, Colorectal polyp prevention by daily aspirin use is abrogated among active smokers. Cancer Causes Control. 2016;27(1):93–103.26510933 10.1007/s 10552-015-0686-1 · doi ↗ · pubmed ↗
- 4Alhammadi N, Asiri AH, Alshahrani FM, Alqahtani AY, Al Qout MM, Alnami RA, Gastrointestinal Complications Associated With Non-steroidal Anti-inflammatory Drug Use Among Adults: A Retrospective, Single-Center Study. Cureus. 2022;14(6):e 26154.35891867 10.7759/cureus.26154 PMC 9302552 · doi ↗ · pubmed ↗
- 5Ricciotti E, Fitz Gerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000.21508345 10.1161/ATVBAHA.110.207449 PMC 3081099 · doi ↗ · pubmed ↗
- 6Nakanishi M, Menoret A, Tanaka T, Miyamoto S, Montrose DC, Vella AT, Selective PGE(2) suppression inhibits colon carcinogenesis and modifies local mucosal immunity. Cancer Prev Res (Phila). 2011;4(8):1198–208.21576350 10.1158/1940-6207.CAPR-11-0188 PMC 3151318 · doi ↗ · pubmed ↗
- 7Nakanishi M, Montrose DC, Clark P, Nambiar PR, Belinsky GS, Claffey KP, Genetic deletion of m PGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008;68(9):3251–9.18451151 10.1158/0008-5472.CAN-07-6100 · doi ↗ · pubmed ↗
- 8Steinmetz-Spah J, Jakobsson PJ. The anti-inflammatory and vasoprotective properties of m PGES-1 inhibition offer promising therapeutic potential. Expert Opin Ther Targets. 2023;27(11):1115–23.38015194 10.1080/14728222.2023.2285785 · doi ↗ · pubmed ↗