Efficient 3D kernels for molecular property prediction
Ankit, Sahely Bhadra, Juho Rousu

TL;DR
This paper introduces efficient 3D graph kernels for predicting molecular properties, improving scalability and accuracy in drug discovery.
Contribution
The 3DGHK kernel reduces computational complexity and outperforms existing 2D/3D kernels and deep learning models.
Findings
3DGHK reduces complexity from O(n6) to O(n2(m+log(n)+δ2+dT6)).
3DGHK outperforms state-of-the-art kernels and deep learning models in classification accuracy.
Experiments on 21 datasets show improved scalability and performance for virtual screening.
Abstract
This paper addresses the challenge of incorporating 3-dimensional (3D) structural information in graph kernels for machine learning-based virtual screening, a crucial task in drug discovery. Existing kernels that capture 3D information often suffer from high computational complexity, which limits their scalability. To overcome this, we propose the 3D chain motif graph kernel, which effectively integrates essential 3D structural properties—bond length, bond angle, and torsion angle—within the three-hop neighborhood of each atom in a molecule. In addition, we introduce a more computationally efficient variant, the 3D graph hopper kernel (3DGHK), which reduces the complexity from the state-of-the-art O(n6) (for the 3D pharmacophore kernel) to O(n2(m+log(n)+δ2+dT6)). Here, n is the number of nodes, T is the highest degree of the node, m is the number of edges, δ is the diameter of the…
Click any figure to enlarge with its caption.
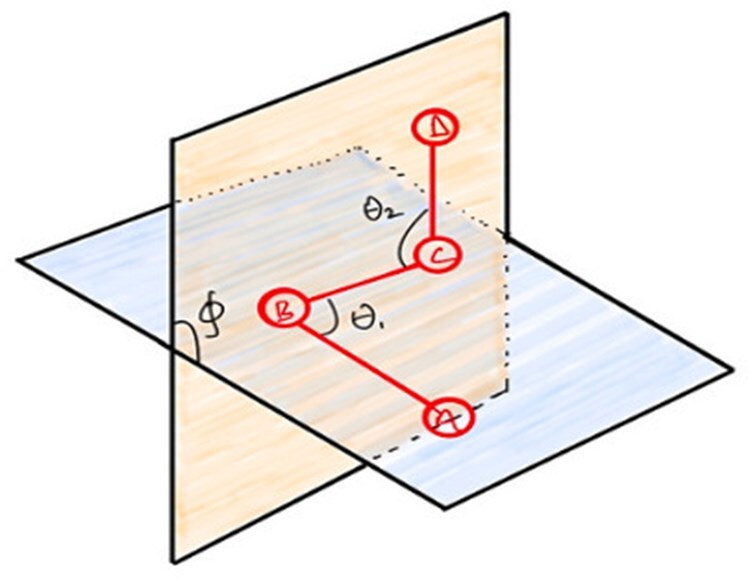 Figure 1
Figure 1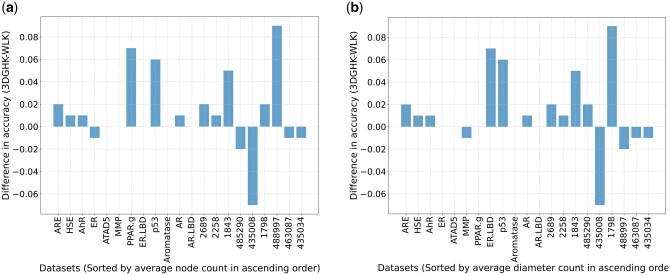 Figure 2
Figure 2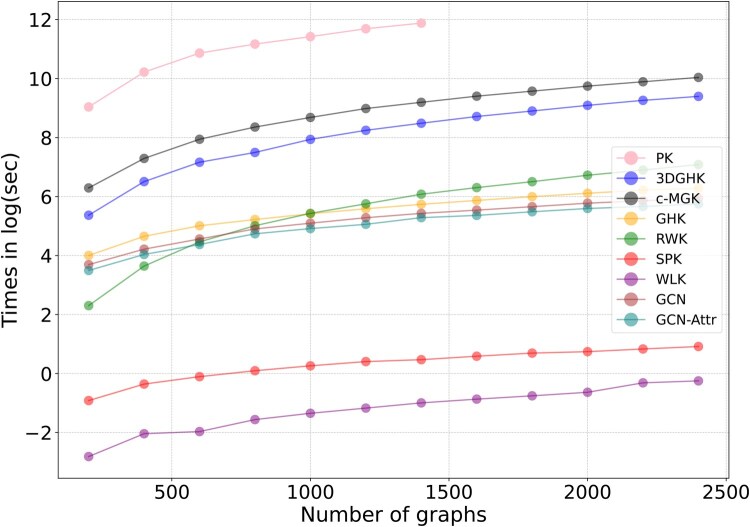 Figure 3
Figure 3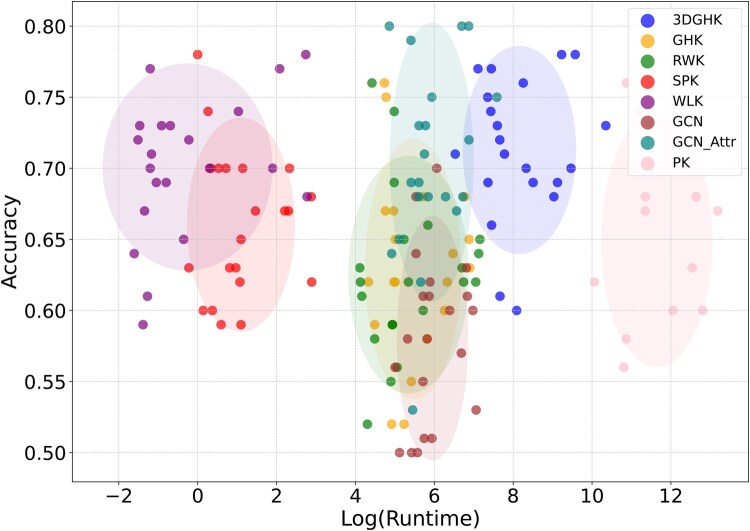 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsComputational Drug Discovery Methods · Protein Structure and Dynamics · Machine Learning in Materials Science