Harlequin Ichthyosis: A Case Report
Shoaib Akhtar, Adeel Anwaar, Inam Ul Haq, Hazaq Mukhtar, Sabir Jamal, Muhammad Muzammil, Aymar Akilimali

TL;DR
This paper reports a case of Harlequin ichthyosis, a rare genetic skin disorder, and highlights the importance of early care and management for better outcomes.
Contribution
The paper contributes a detailed case report emphasizing the need for multidisciplinary care in managing Harlequin ichthyosis.
Findings
Early recognition and neonatal care are crucial for improving survival in Harlequin ichthyosis.
Multidisciplinary management helps in effectively managing symptoms and enhancing quality of life.
Treatment strategies focus on hydration, infection prevention, and supportive care.
Abstract
Harlequin ichthyosis (HI) is a genetic disorder caused by ABCA12 gene mutations, presenting with thick, scaly skin and deep fissures. Early recognition, intensive neonatal care, and multidisciplinary management are crucial for improving survival and quality of life. Treatment focuses on skin hydration, infection prevention, and supportive care to manage symptoms effectively.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
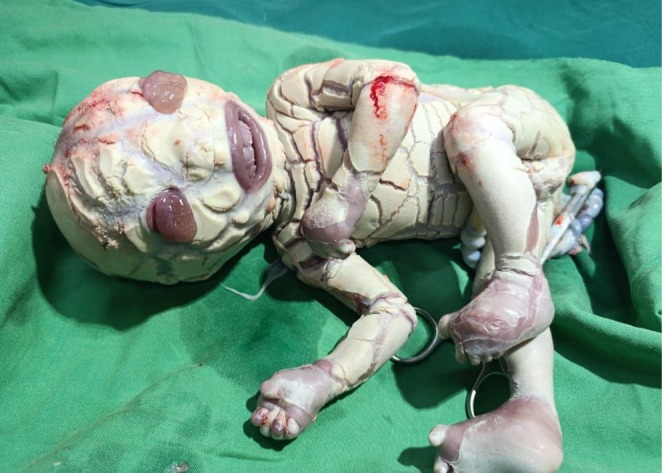 FIGURE 1
FIGURE 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSkin and Cellular Biology Research · Mast cells and histamine · Polyamine Metabolism and Applications
Introduction
1
Harlequin ichthyosis (HI) is one of the most severe forms of congenital ichthyosis, a group of disorders characterized by abnormal skin shedding and keratinization. It is an autosomal recessive condition resulting from mutations in the ABCA12 gene [1, 2, 3], which encodes a transporter protein responsible for lipid transport in the epidermis [4]. This disruption leads to the accumulation of thick scales and the formation of deep, painful fissures in the skin, often leading to systemic complications such as dehydration, electrolyte imbalances, and infections. The classic “harlequin” appearance of the skin—marked by large, diamond‐shaped scales—gives the disorder its name. While the condition was once associated with a high mortality rate, advances in neonatal care and treatment strategies have improved the prognosis for affected individuals. We report a rare case of HI.
Case Presentation
2
Case History/Examination
2.1
A 21‐year‐old pregnant woman was admitted to Punjab Rangers Teaching Hospital, Lahore, for her second pregnancy due to preterm labor and lower abdominal pain (obstetric pain). The gestational age was approximately 32 weeks based on both the first day of her last menstrual period and ultrasound findings.
Imaging Investigation Findings
2.2
Polyhydramnios was observed on ultrasound, with an amniotic fluid index of 26, indicating an excessive accumulation of amniotic fluid. The underlying cause of the condition remained uncertain, as no further diagnostic evaluations had been conducted. Notably, an anomaly scan had not been performed, leaving potential fetal abnormalities unassessed. Further investigations were needed to determine any underlying maternal or fetal factors contributing to the increased fluid levels.
Surgical Management
2.3
A male baby with HI was delivered via lower‐segment cesarean section, highlighting the complexities involved in his perinatal management. His birth weight, occipitofrontal circumference, and length were recorded as 1.56 kg, 31 cm, and 44 cm, respectively, and these measurements indicated that he was significantly small, underscoring his fragile condition. Notably, the physical examination revealed features such as thick skin with deep fissures, generalized hyperkeratinization, cyanosis, flat fontanels, ectropion, immature eyes and auricles, eclabium, and moaning (Figure 1), which collectively emphasized the severity of his congenital condition, while the fact that his parents, who were distantly related, already had one healthy child added an intriguing aspect to his genetic background.
A baby boy with deep cracked skin, a wide‐open mouth, abnormal eyes, and a flattened nose and ear.
Ongoing Follow‐Up Plan
2.4
Antibiotic therapy and conservative treatments were initiated following admission to the neonatal intensive care unit to manage infections and provide supportive care. Despite intensive medical interventions, the baby's condition remained critical due to the severity of HI and associated complications. Unfortunately, his health continued to deteriorate, and he succumbed to his condition after 14 days of life. His passing highlighted the challenges in managing this rare and severe genetic disorder, despite advances in neonatal care.
Discussion
3
The presentation of HI is typically immediate at birth, with infants displaying thick, rigid skin, deep fissures, and erythematous areas due to the formation of large, diamond‐shaped scales. The skin often appears cracked and tense, impairing the infant's ability to move and breathe effectively. Other manifestations include ear and eye deformities, as well as difficulty with thermoregulation and feeding [5].
HI results from mutations in the ABCA12 gene [1, 2, 3], which leads to defective lipid transport in the epidermal cells, causing defective barrier function [1, 4]. This defect leads to the excessive accumulation of keratinocytes, which form thick, adherent plaques that impair normal skin shedding [6].
The management of HI is primarily supportive, with the goal of preventing infections, maintaining hydration, and managing pain [6, 7]. Early interventions include the use of emollients and keratolytic agents to soften the scales and encourage their removal. Intravenous fluids and electrolytes may be required to correct imbalances caused by skin barrier dysfunction [8]. Antibiotic treatment is often necessary to prevent or treat infections, and respiratory support may be needed to ensure adequate oxygenation. Given the severity of the condition, early multidisciplinary care is crucial to improving outcomes. The involvement of dermatologists, neonatologists, physical therapy specialists, an orthopedic team, plastic surgeons, and pediatric specialists is key for providing comprehensive care [9].
Advances in neonatal intensive care have improved the survival rate of infants with HI. Many infants, once considered unlikely to survive beyond the first few days, can now live into childhood with appropriate care [10]. Long‐term management focuses on skin care, infection prevention, and addressing any systemic complications [5, 6].
Although life expectancy has improved, patients often face lifelong dermatological and psychosocial challenges.
Conclusion and Results
4
HI remains a challenging condition for both patients and clinicians, requiring early recognition and a multidisciplinary management approach. Advances in neonatal care have significantly improved survival rates, but the condition requires ongoing management throughout life. Early intervention, particularly in the neonatal period, is critical to reducing morbidity and mortality. Genetic counseling and prenatal diagnosis may be useful for families with a known risk of the condition.
Author Contributions
Shoaib Akhtar: conceptualization, supervision, validation, visualization, writing – original draft, writing – review and editing. Adeel Anwaar: project administration, supervision, validation, visualization, writing – original draft, writing – review and editing. Inam Ul Haq: data curation, visualization, writing – original draft, writing – review and editing. Hazaq Mukhtar: visualization, writing – original draft, writing – review and editing. Sabir Jamal: writing – original draft, writing – review and editing. Muhammad Muzammil: writing – original draft, writing – review and editing. Aymar Akilimali: validation, visualization, writing – original draft.
Ethics Statement
This is a case report utilizing anonymized patient information and so was classified as exempt from review from the institutional review board.
Consent
A written informed consent was obtained from the patient based on the journal's policies.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1V. Oji , “Ichthyosis:Clinical Genetics and Pathophysiology,” Journal of Investigative Dermatology 130, no. 6 (2010): 1423–1432.
- 2F. J. D. Smith , “Mutations in the ABCA 12 Gene Are Associated With Harlequin Ichthyosis,” Nature Genetics 38, no. 3 (2006): 376–381.
- 3N. V. Whittock , “Harlequin Ichthyosis and Other Congenital Ichthyoses: The Molecular and Genetic Basis,” British Journal of Dermatology 137, no. 1 (1997): 1–5.9274618
- 4A. Hovmanian , “Harlequin Ichthyosis Unmasked: A Defect of Lipid Transport,” Journal of Clinical Investigation 115 (2005): 1708–1710.16007249 10.1172/JCI 25736 PMC 1159155 · doi ↗ · pubmed ↗
- 5S. Rajpopat , C. Moss , J. Mellerio , et al., “Harlequin Ichthyosis: A Review of Clinical and Molecular Findings in 45 Cases,” Archives of Dermatology 147, no. 6 (2011): 681–686.21339420 10.1001/archdermatol.2011.9 · doi ↗ · pubmed ↗
- 6M. Akiyama , “Harlequin Ichthyosis: Pathogenesis and Clinical Management,” Journal of Dermatology 43, no. 5 (2016): 477–484.
- 7C. Munns , “A Comprehensive Review of the Clinical Care of Children With Harlequin Ichthyosis,” Pediatric Dermatology 34, no. 1 (2017): 57–63.
- 8A. Shibata and M. Akiyama , “Epidemiology, Medical Genetics, Diagnosis and Treatment of Harlequin Ichthyosis in Japan,” Pediatrics International 57, no. 4 (2015): 620–626.25857373 10.1111/ped.12638 · doi ↗ · pubmed ↗