Unraveling Dowling–Degos Disease: A Rare Skin Disorder
Mahesh Mathur, Neha Thakur, Supriya Paudel, Sambidha Karki, Sandhya Regmi, Nabita Bhattarai

TL;DR
This paper presents a case of a rare skin disorder called Dowling–Degos disease in a 51-year-old woman and her family.
Contribution
The study reports a familial case of DDD with clinical and histopathological confirmation.
Findings
The patient had asymptomatic brownish-black reticular lesions in flexural areas.
The patient's family members also showed similar skin lesions.
The diagnosis was confirmed through clinical and histopathological evaluation.
Abstract
Dowling–Degos disease (DDD) is a rare genodermatosis characterized by brown to black macules distributed symmetrically in the axilla, groin, elbow, face, neck, and trunk. It is more common in women, usually after puberty. The main pathogenesis behind DDD is a mutation in the keratin 5 gene. Here, we present a case of 51‐year‐old female presenting as asymptomatic brownish‐black lesions arranged in a reticular pattern involving flexural sites. The clinical and histopathological findings are consistent with DDD. Her mother, brother, son, and daughter also had similar lesions. The patient was counseled about the prognosis and treatment options of the disease.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
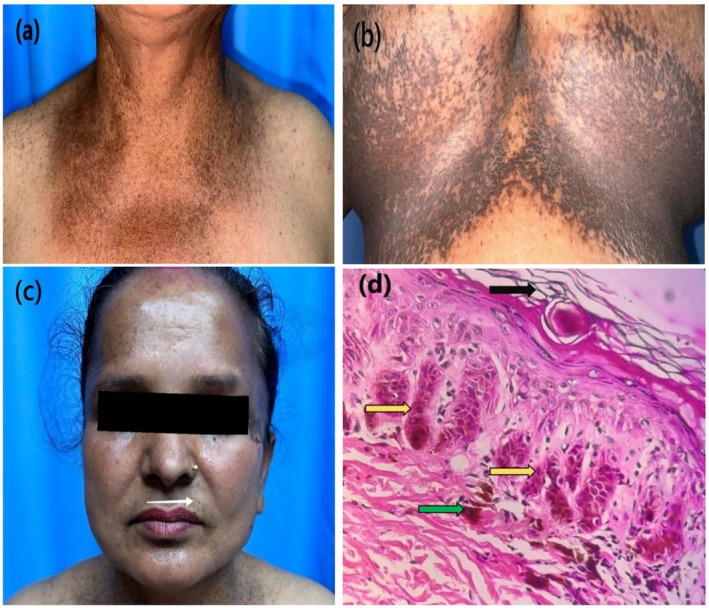 FIGURE 1
FIGURE 1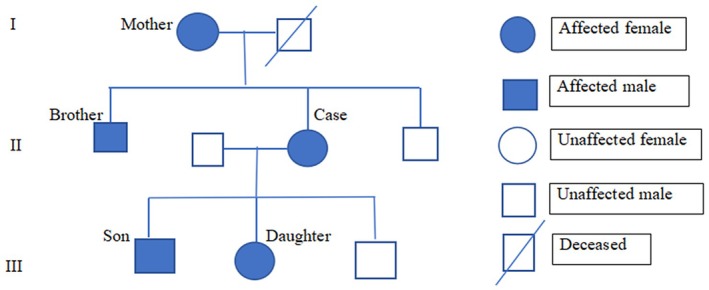 FIGURE 2
FIGURE 2| Differential diagnosis | Differentiating features |
|---|---|
| Galli‐Galli disease | Reticulated hyperpigmentation in flexures but supra basal dyskeratotic acantholysis in HPE |
| Reticulate acropigmentation of Kitamura | Childhood onset, atrophic hyperpigmented papules coalescing into a reticulated pattern in acral areas |
| Haber syndrome | Photosensitive rosacea‐like facial eruptions initially followed by keratotic papules, comedo‐like lesions, pitted scars, reticulate hyperpigmentation on trunk, proximal extremities and axillae |
| Dyschromatosis symmetrica hereditaria | Pinpoint, hypo and hyperpigmented macules on dorsal aspects of the hand and feet |
| Dyschromatosis universalis hereditaria | Pigmented flecks and spots on body parts other than flexures |
| Neurofibromatosis type 1 | Axillary and inguinal freckling with multiple neurofibromas and other cutaneous manifestations |
| Acanthosis nigricans | Velvety plaques, less elongation of rete ridges and no follicular involvement in HPE |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRNA regulation and disease · RNA Research and Splicing · Acne and Rosacea Treatments and Effects
Summary
- Dowling–Degos disease (DDD) is an uncommon autosomal dominant disorder characterized by reticulate pigmentation.
- It is important to diagnose the disease as it mimics many other cutaneous conditions.
- Typical histopathology is pathognomic and aids in the diagnosis of this rare entity.
Introduction
1
Dowling–Degos disease (DDD) is a rare, progressive, autosomal dominant disorder characterized by symmetrical, pigmented macules involving the axillae, groins, face, neck, arms, and trunk as well as scattered comedo‐like lesions and pitted acneiform scars [1]. Its prevalence is not well documented in the literature. DDD was described initially by Dowling and Freudenthal in 1938 [2]. Typically, the onset of DDD is after puberty and commonly occurs in the third to fourth decade of life [3]. Women are more commonly affected than men [2]. Histopathological examination (HPE) of skin lesions is diagnostic. We hereby report a case of DDD considering the rarity of this skin condition.
Case Presentation
2
A 51‐year‐old female presented with multiple asymptomatic reticular hyperpigmented macules, which appeared first on the flexor aspect of the elbow and progressed to involve other flexural parts of the body (Figure 1a,b). Skin lesions were present around the age of 25, and the pigmentation has worsened over the past 4 years. There is a history of similar lesions in her mother, brother, son, and daughter (Figure 2 pedigree chart). On examination, there were multiple brown to black colored macules of 2–4 mm in diameter arranged in a reticular pattern distributed symmetrically over the face, chest, inframammary region, axilla, elbow, and groin. Few pitted scars (Figure 1c) were present in the perioral regions.
(a) Multiple reticular pigmentations over neck and chest. (b) Over inframammary region. (c) Few pitted scars above the upper lip in left side (white arrow). (d) Histopathology: Hematoxylin and eosin staining (40×) with hyperkeratosis (shown by black arrow), “antler‐like” pattern (shown by yellow arrow) and basal hyperpigmentation (shown by green arrow).
Pedigree of the family with Dowling–Degos disease.
Methods
3
Routine laboratory investigations were within normal limits. HPE of skin biopsy from right inframammary region showed hyperkeratosis, orthokeratosis, focal parakeratosis, and thinning of suprapapillary epidermis. The epithelial strands extend into the superficial dermis from the epidermis and hair follicles resulting in an “antler‐like” pattern (Figure 1d). Basal hyperpigmentation and mild perivascular lymphocytic infiltration in dermis were also noted.
Results
4
On the basis of clinical and histological findings, the diagnosis of DDD was made. The patient was counseled about the prognosis of the disease and treatment options.
Discussion
5
DDD is a rare autosomal dominant genodermatosis but can also occur sporadically. DDD is clinically characterized by asymptomatic, progressive, reticulate hyperpigmented macules with a flexural distribution. Other cutaneous findings include comedo‐like hyperkeratotic follicular papules on the neck and perioral pitted acneiform scars [4]. In our case, classical reticulated pigmentations were present in flexures; however, comedo‐like papules were not seen.
Loss‐of‐functional mutation in the keratin 5 (KRT5) gene on chromosome 12q13 is reported. Mutation of this gene results in disruption in melanosome transfer and trafficking, disrupting the normal skin pigmentation and structure [5]. Other genes involved in pathogenesis are POGLUT1, POFUT, PSENEN, and DSRAD gene in chromosome 17p13.3 and chromosome 1q21 [1, 5]. Genetic studies are not carried out in our case due to affordability issues.
Epidermal cysts, multiple keratoacanthomas, squamous cell carcinoma, abscess, hidradenitis suppurativa, seborrheic keratosis, and pilonidal cysts are associated with DDD [2]. These disorders are not present in our case. Histopathology of the skin biopsy in classical DDD reveals thinning of the epidermis, elongated, thinned, and branched (antler‐like) rete ridges with increased melanin pigmentation at their tips, but no increase in the melanocyte number [6]. Histopathology of the skin biopsy of our patient also revealed these classical findings.
Variety of hyperpigmentation disorders appearing in flexural areas that should be differentiated from DDD are Galli–Galli disease, reticulate acropigmentation of Kitamura, Haber syndrome, dyschromatosis symmetrica hereditaria, dyschromatosis universalis hereditaria, neurofibromatosis type 1, and acanthosis nigricans. Details regarding these differential diagnoses are described in Table 1 [7, 8, 9]. This case report highlights the importance of recognizing the clinical and histopathological features of DDD for accurate diagnosis and differentiation from other similar conditions.
There is no definitive cure for DDD. Various treatment options have been tried in recent years, but none of them have shown satisfactory results. Treatment options include depigmenting agents such as hydroquinone, retinoids, and laser therapies such as fractional erbium YAG laser. Despite these options, effective and long‐term solutions remain difficult to achieve [10].
Conclusions
6
DDD is an uncommon and non‐life‐threatening genetic disorder. However, discoloration associated with DDD can lead to psychological distress. Continued research and documentation of such cases may lead to better treatment options in the future.
Author Contributions
Mahesh Mathur: conceptualization, formal analysis, resources, supervision, validation, visualization, writing – original draft. Neha Thakur: conceptualization, formal analysis, resources, supervision, validation, visualization, writing – original draft. Supriya Paudel: formal analysis, resources, supervision, visualization, writing – original draft, writing – review and editing. Sandhya Regmi: data curation, investigation, resources, visualization, writing – review and editing. Sambidha Karki: data curation, investigation, resources, visualization, writing – review and editing. Nabita Bhattarai: conceptualization, formal analysis, resources, supervision, validation, visualization, writing – original draft.
Ethics Statement
Reviewed and approved by Institutional review board College of medical sciences (IRBCOMS). The patients in this manuscript have given written informed consent to the publication of their case details.
Consent
The authors obtained written consent from the patient for use of photographs and medical information to be published online and with the understanding that this information may be publicly available and discoverable via search engines. Patient consent forms are not provided to the journal but are retained by the authors.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1J. Wititsuwannakul and N. Noppakun , “Generalized Dowling‐Degos Disease: Case Reports,” Annals of Dermatology 25, no. 3 (2013): 360–364.24003282 10.5021/ad.2013.25.3.360PMC 3756204 · doi ↗ · pubmed ↗
- 2S. G. Rathoriya , S. S. Soni , and D. Asati , “Dowling‐Degos Disease With Reticulate Acropigmentation of Kitamura: Extended Spectrum of a Single Entity,” Indian Dermatology Online Journal 7, no. 1 (2016): 32–35.26955585 10.4103/2229-5178.174307 PMC 4763577 · doi ↗ · pubmed ↗
- 3A. S. Rice and C. Cook , “Dowling‐Degos Disease,” in Stat Pearls [Internet] (Stat Pearls Publishing, 2024).30285365 · pubmed ↗
- 4R. Bachaspatimayum , N. Bhattacharjee , and P. Das , “Dowling Degos Disease in a Child: A Rare Case Report From Northeast India,” Indian Journal of Paediatric Dermatology 22, no. 3 (2021): 264–266.
- 5C. Stephan , M. Kurban , and O. Abbas , “Dowling‐Degos Disease: A Review,” International Journal of Dermatology 60, no. 8 (2021): 944–950, 10.1111/ijd.15385.33368260 · doi ↗ · pubmed ↗
- 6V. Kharayat , P. Sinha , M. G. Madakshira , and V. Sharma , “Dermoscopy of Classic Dowling–Degos Disease With Review of Literature,” Pigment International 10 (2023): 60–63.
- 7S. Şengiz Erhan , T. Yüksel , and P. Engin Zerk , “Dowling‐Degos Disease: A Case Report,” European Archives of Medical Research 37, no. 1 (2021): 52–55.
- 8E. F. Georgescu , L. Stanescu , C. F. Popescu , M. Comanescu , and I. Georgescu , “Dowling‐Degos Disease,” Romanian Journal of Morphology and Embryology 51, no. 1 (2010): 181–185.20191141 · pubmed ↗