Cytokine signaling defects in primary atopic diseases—an updated review
Vaishali Thakur, Rakesh Kumar Pilania, Arunima Sharma, Saniya Sharma, Alfred Thomas Mario, Taru Goyal, Madhubala Sharma, Gayathri Coimbatore Vaitheeswaran, Pandiarajan Vignesh, Surjit Singh, Manpreet Dhaliwal, Amit Rawat

TL;DR
This review discusses how defects in cytokine signaling pathways contribute to primary atopic diseases, emphasizing the importance of early diagnosis for better treatment.
Contribution
The paper provides an updated review of cytokine signaling defects in primary atopic diseases, highlighting key pathways and their clinical implications.
Findings
Defects in STAT3, JAK1/STAT5b, and TGF-β pathways are key in primary atopic diseases.
Pathogenic variants in these pathways lead to Th2 polarization and severe atopic symptoms.
Early differentiation between primary atopic diseases and polygenic atopy is crucial for targeted interventions.
Abstract
Primary atopic disorders (PADs) are monogenic conditions associated with severe, early-onset atopic diseases. Clinically, they often overlap with polygenic allergic conditions, making specialized laboratory testing necessary to distinguish them from polygenic atopy. Multisystem involvement, such as growth failure, recurrent infections, and autoimmunity, points towards PADs warranting further investigations. PADs associated with immune dysregulation can be broadly categorized into four mechanistic groups: those affecting the regulation of cell cytoskeleton dynamics, T-cell receptor (TCR) signaling and repertoire diversity, and function of regulatory T cell (Treg), and cytokine signaling. In this review, we have examined the defects in cytokine signaling pathways associated with PADs. Key cytokine signaling pathways implicated in PADs include the STAT3, JAK1/STAT5b, and TGF-β pathways.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
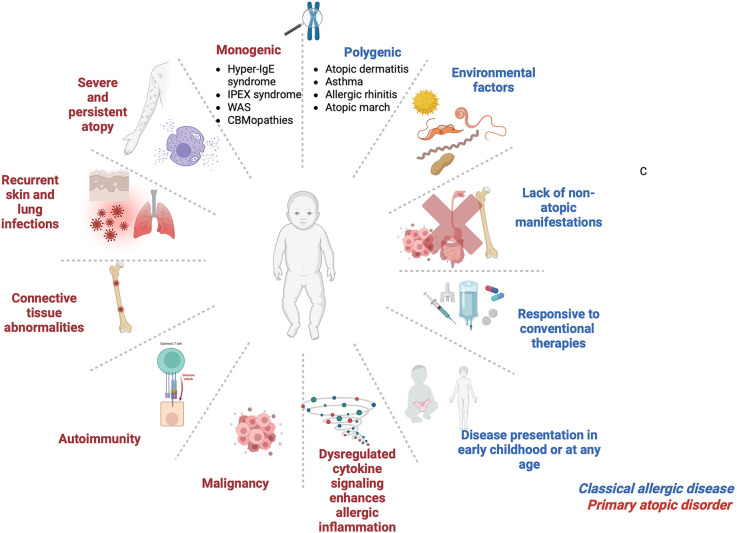 Figure 1
Figure 1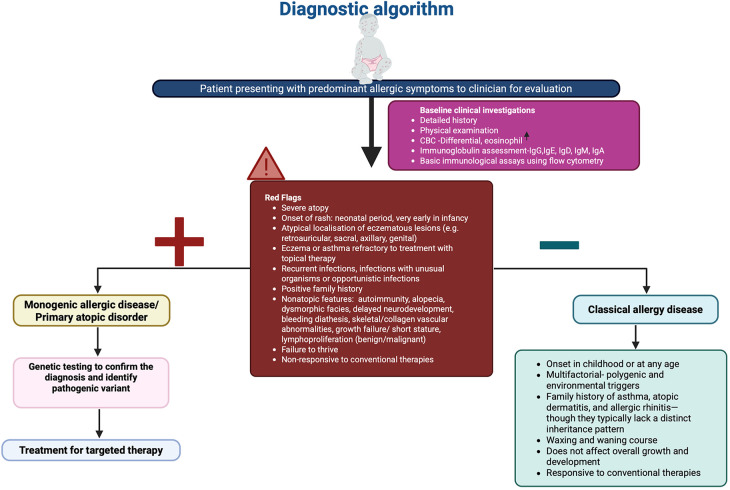 Figure 2
Figure 2| S.No. | Gene | GOF/LOF molecular consequences | OMIM genotype no | OMIM phenotype | Inheritance pattern | Associated clinical phenotype | Targeted therapy |
|---|---|---|---|---|---|---|---|
| 1 |
| GOF | *600555 | 614162 | AD | CMC, autoimmunity, type I interferonopathies, IPEX-like syndrome | Ruxolitinib, ( |
| 2 |
| DN | *102582 | 147060 | AD | HIES, eczema, recurrent skin and lung infections, eosinophilia, candidiasis, skeletal and connective tissue abnormalities retained primary teeth, characteristic facies, cerebral aneurysms | Tocilizumab and JAK inhibitors, ( |
| 3 |
| GOF | *604260 | AR-245590 | Somatic | Hypereosinophilic syndrome, growth failure, immune dysregulation, and lymphoproliferation. | JAK inhibitors (Ruxolitinib ( |
| 4 |
| LOF | *604260 | AD, AR | Eczema, IPEX-like autoimmune manifestations Autosomal dominant form causes dermatitis but without severe immunodeficiency | Cyclosporine therapy ( | |
| 5 |
| GOF | *601512 | 620532 | AD | Severe and treatment-resistant dermatitis, marked eosinophilic gastrointestinal disease | Ruxolitinib Tofacitinib Dupilumab ( |
| 6 |
| LOF | *618269 | 618282 | AR | Phenocopy of STAT3 DN | Dupilumab ( |
| 7 |
| LOF | *147730 | 606367 | AR | IPEX-like syndrome (e.g., enteropathy, endocrinopathies, and failure to thrive) | Rapamycin ( |
| 8 |
| GOF | *147781 | AD | Early-onset atopic dermatitis, hyper IgE levels, food allergies, asthma and autoimmunity | Dupilumab ( | |
| 9 | LOF | *600694 | 619752 | AD | Phenotypic overlap with AD-HIES Stuve-Wiedemann-like syndrome | Supportive treatment ( | |
| 10 |
| LOF | *147880 | 618944 | AR | Partially overlapping with AD-HIES: No connective tissue abnormalities | Immunoglobulin replacement therapy ( |
| 11 |
| LOF | *190181 | 609192 | AD | Marfan-like syndrome; phenotypic overlap with STAT3 pathway disorders | HSCT surgical interventions ( |
| 12 |
| GOF | *147795 | 618999 | AD | Hypereosinophilic syndrome | JAK inhibitors like ruxolitinib and tofacitinib ( |
| 13 |
| LOF | *606944 | AD | Significant eosinophilic esophagitis, cutaneous mastocytosis, connective tissue abnormalities | Dupilumab ( |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsImmune Cell Function and Interaction · T-cell and B-cell Immunology · IL-33, ST2, and ILC Pathways
Introduction
1
Inborn errors of immunity (IEIs) are heritable disorders with a heterogenous presentation due to variants in genes impairing the activity of the immune system (1). Clinically, they may manifest as heightened susceptibility to infections, autoinflammation, autoimmunity, atopy, and malignancies (2). A subset of these IEIs features a distinct atopic phenotype marked by chronic Th2 skewing, aberrant mast cell degranulation, eosinophilic inflammation, and elevated IgE levels (3). In 2018, Lyons and Milner introduced the term primary atopic disorders (PADs) to categorize monogenic conditions associated with early-onset atopic symptoms driven by immune dysregulation (4). However, it is now recognized that PADs are not a strict subcategory of IEIs. Only a subset of PADs are immune in origin and may be classified as IEI. Many PADs result from non-immune mechanisms involving structural or barrier defects. These PADs primarily disrupt epithelial integrity, leading to heightened allergen penetration and subsequent atopic responses. Prototypic disorders of these PADs are attributed to variants in genes encoding epidermal barrier proteins like FLG (filaggrin), protease inhibitors such as SPINK5, and intercellular adhesion molecules CDSN, DSG1, and DSP (5–7).
IEIs with atopic manifestations exhibit overlapping clinical and immunological phenotypes and have been grouped into key syndromic categories. These are classified based on clinical presentation and immunological profiling as Hyper-IgE syndromes, immune dysregulation poly-endocrinopathy enteropathy X-linked (IPEX) and IPEX-like conditions, Omenn syndrome, Wiskott–Aldrich syndrome, CBM-opathies, and other atopy predominant IEIs (8). Although clinical features of PADs often overlap with polygenic allergic conditions, they are distinguished by early onset and severe manifestations with complex comorbidities such as growth failure, recurrent infections, and autoimmunity, to name a few (9) (Figure 1).
Clinical characteristics for classical allergic disease and primary atopic disorder. IPEX, Immune dysregulation, polyendocrinopathy, enteropathy, X-linked; WAS, Wiskott–Aldrich syndrome; CBMopathies, CBM complex—CARD11, BCL10 and MALT1. Figure was created using BioRender.
Atopy is a genetic tendency to mount exaggerated IgE-mediated immune responses to environmental allergens. These patients often present with a constellation of clinical symptoms, such as atopic dermatitis, food allergy, allergic rhinitis, and asthma - collectively referred to as the ‘atopic march’ (10–13). In PADs, these manifestations are often accompanied by immune dysregulation due to Th2 polarization and overproduction of IL-4, IL-5, and IL-13. These Th2 effector cytokines drive downstream signaling cascades that recruit eosinophils, mast cells, and other effector cells (11). While allergic phenotypes are common in the general population, those seen in IEIs are frequently more severe and are rooted in well-defined genetic defects (9).
Diagnosing PADs begins with a thorough clinical evaluation, including physical examination, family history, and immunological investigations (14) (Figure 2). Family medical history is important as PADs typically exhibit distinct inheritance patterns, although de-novo variants may arise spontaneously as well. Hallmark laboratory findings include eosinophilia, elevated serum IgE, abnormal immunoglobulin profiles, and T-cell subsets, which are typically assessed via flow cytometry (14). Genetic testing plays a pivotal role in confirming the diagnosis by identifying causal variants (15).
Diagnostic algorithm in patients with suspected primary atopic disorder. Figure was created using BioRender.
Key immune pathways involved include those regulating the cellular cytoskeleton along with immune synapse formation, T-cell receptor (TCR) signaling and repertoire diversity, T regulatory cell (Treg) function, and innate immune cell effector mechanisms (16). Among the molecular mechanisms implicated in PADs, signaling pathways of cytokines have a cardinal role. Disruption in cytokine signaling can severely impair host immune response and tolerance, predisposing individuals to infections, inflammation, and allergic diseases (17). Specifically, alterations in STAT3, JAK1/STAT5, and TGF-β pathways through molecular consequences, such as dominant-negative (DN), loss or gain-of-function (LOF, GOF) mutations, are closely associated with Th2 polarization and atopic manifestations (14).
In this review, we focus on PADs arising from genetic defects that disrupt cytokine signaling networks. We examine mutations affecting transcription factors such as STAT1, STAT3, STAT5B, STAT6, ZNF341, cell surface receptors including IL2RA, IL4RA, IL6R, IL6ST, TGFBR1/2, and intracellular signaling mediators such as JAK1 and ERBB2IP (18). Understanding these pathways not only enhances our insight into the pathogenesis of PADs but also provides a foundation for developing precision medicine. The key primary atopic disorders resulting from cytokine signaling defects have been summarised in Table 1.
Role of transcription factors in altered cytokine signaling
2
STAT1 GOF
2.1
Autosomal dominant- GOF variants in STAT1 are among the commonest monogenic defects linked to chronic mucocutaneous candidiasis (CMC), with over 400 reported cases (19, 20). These mutations prevent dephosphorylation of STAT1, leading to its constitutive nuclear localization and enhanced type I/II interferon signaling (21). Consequently, Th17 differentiation is impaired, which directly impacts antifungal defenses, and enhanced interferon signaling drives autoimmunity (e.g., hypothyroidism, cytopenias) that resemble IPEX-like phenotypes (22). A novel N-terminal mutation (c.194A>C; p.D65A) has even been tied to eosinophilic esophagitis, underscoring the link between STAT1 hyperactivity and atopic inflammation (21, 23).
STAT3 DN
2.2
First described clinically as Job's syndrome in 1966, autosomal -dominant DN mutations in STAT3 are the molecular basis of Hyper -IgE syndrome (HIES) (24–26). Patients exhibit severe eczema, elevated IgE (>1,000 IU/ml), eosinophilia, and recurrent staphylococcal skin and pulmonary infections (27, 28). Dampened IL-6 and IL-10 signaling through STAT3 reduces Th17 cell numbers and IL-17 production, heightening susceptibility to Staphylococcus and Candida infections (27, 29). Diagnostic criteria combine IgE quantification, Th17 enumeration, a clinical scoring system (>30 points), and genetic confirmation of a STAT3 DN variant (30).
STAT5B GOF
2.3
Somatic GOF variants in the SH2 or transactivation domains of STAT5B enhance STAT5 signaling, driving clonal T-cell expansion with a Th2 bias (31). Thus, STAT5B remains constitutively active instead of responding appropriately to growth hormone signals that regulate IGF-1–dependent growth but also skew T-cell differentiation towards a Th2 phenotype. Clinically, affected individuals present with treatment-refractory atopic dermatitis, persistent urticaria, elevated numbers of eosinophilia, alopecia, and angioedema (32).
STAT5B LOF
2.4
STAT5B loss-of-function leads to atopic dermatitis with dwarfism, hyper IgE, autoimmunity and lymphocytic interstitial pneumonitis (33, 34). STAT5B acts as a key mediator in growth hormone signaling, and its deficiency results in growth hormone insensitivity and growth failure (35). Additionally, STAT5B LOF impairs IL-2-dependent signaling, leading to recurrent viral infections associated with reduced function and even decreased numbers of T regs (34). Atopic symptoms such as eczema are prevalent and affected individuals may develop conditions resembling IPEX like syndrome (33). In addition to LOF variants, the STAT5B deficiency can also result from the autosomal dominant form of STAT5B, causing stunted growth and eczema, but it does not lead to severe immunodeficiency (36).
STAT6 GOF
2.5
STAT6 is the major transcription factor activated by IL-4 and IL-13. Upon activation, STAT6 dimerizes and translocates to the nucleus. STAT6 promotes differentiation of naive CD4^+^ T cells to Th2 cells along with class-switch from IgM to IgE on B-cells (37). This process is initiated on binding of IL-4 and IL-13 to the IL-4 receptor complex, triggering the phosphorylation of tyrosine residues on the IL-4 receptor alpha (IL-4Rα) subunit (38). This is mediated via Janus kinases (JAK) (39). Src homology 2 (SH2) domains recruit STAT6, which bind to the phosphorylated tyrosine residues on IL-4Rα (39). In STAT6 GOF, hyperphosphorylation of STAT6 intensifies IL-4 and IL-13 signaling. A STAT6 heterozygous misense variant (c.1129G>A; p.Glu377Lys) linked to atopy was initially identified by Suratannon et al. (40). Another missense variant in exon 22 (c.1114G>A; p.E372K) was identified in a patient with early-onset eczema, food allergies, eosinophilia, and eosinophilic esophagitis (41). Functional studies demonstrated heightened IL-4/IL-13 responsiveness, reversed by JAK inhibition (ruxolitinib) or IL-4Rα blockade, which normalized IgE levels and tissue eosinophilia (41). To date, STAT6-GOF mutations have been reported in 21 persons (42).
ZNF341 LOF
2.6
ZNF341 is a zinc-finger transcription factor that upregulates both STAT1 and STAT3 expression (43). Autosomal-recessive LOF mutations in ZNF341 phenocopy STAT3-HIES, causing elevated IgE, eosinophilia, eczema, and recurrent bacterial and fungal infections (43, 44). Till now, 20 patients with autosomal recessive LOF ZNF341 have been reported (44). Unlike STAT3 DN, connective tissue defects tend to be milder, but the underlying mechanism, diminished STAT3 transcription remains the same.
Cell surface receptor defects in altered cytokine signaling
3
IL-2Rα (CD25) LOF
3.1
The α-chain of the high-affinity IL-2 receptor is encoded by IL2RA and is essential for regulatory T-cell development and peripheral tolerance (45). Biallelic loss-of-function mutations in IL2RA produce an IPEX-like syndrome characterized by severe atopic dermatitis, eosinophilia, elevated IgE, autoimmunity, and chronic infections (22, 46). Defective IL-2 signaling impairs Treg homeostasis, which results in unchecked Th2 and Th17 responses that drive both allergic and autoimmune pathology (22).
IL-4RA GOF
3.2
Gain-of-function (GOF) variants in IL4RA, particularly the R576 allele, are strongly associated with increased susceptibility to atopic diseases (47). The Q576R variant in IL-4RA disrupts the formation of the STAT3–ERBIN–SMAD2/3 complex (48). Impaired STAT3 and ERBIN function intensifies Th2 polarization by reducing TGF-β signaling and increasing IL-4RA expression on lymphocytes (49). These changes culminate in a clinical phenotype characterised by early-onset atopic dermatitis, elevated serum IgE, asthma, food allergies, and, in some cases, autoimmune features (50). Importantly, dupilumab, an IL-4Rα antagonist, has demonstrated clinical efficacy in treating patients with variants in IL-4RA (51).
IL6ST LOF
3.3
IL6ST encodes GP130, the shared signal-transducing subunit for all IL-6 family cytokines. The main cytokines of IL-6 family include IL-6, IL-11, IL-27, IL-35, IL-39, and oncostatin M (52, 53). Recessive LOF variants abolish JAK/STAT3 activation, manifesting as an autosomal-recessive Hyper-IgE syndrome with eczema, high IgE, eosinophilia, and recurrent bacterial infections (54). Recently, dominant-negative IL6ST mutations (c.2261C>A, p.Ser754Ter) have been linked to autosomal-dominant HIES phenotypes (54, 55), and secondary glycosylation defects (e.g., in PGM3 deficiency) can similarly impair GP130 surface expression and STAT3 phosphorylation (56).
IL-6R LOF
3.4
One of the key functions of IL-6 signaling is to differentiate activated Th cells into IL-17 and IL-22 secreting Th17 and Th22 cells (53).). On the other hand, IL-6 suppresses the differentiation of CD4+ T regulatory cells, which regulate inflammation. Individuals deficient in IL-6R develop atopic dermatitis, eosinophilia, recurring pulmonary infections, skin abscesses due to Staphylococcus sp., high IgE levels, but no skeletal abnormalities (57, 58).
TGF-β receptor (TGFBR1/2) deficiency in Loeys–Dietz syndrome
3.5
Heterozygous mutations in TGFBR1 or TGFBR2 cause Loeys–Dietz syndrome (59). This is an autosomal-dominant connective-tissue disorder marked by arterial aneurysms, craniofacial abnormalities, and severe atopic features in the form of asthma, food allergy, and eosinophilic gastrointestinal disease (60). The TGFBR1/2 complex recognises TGF-β, and variants in the receptor may lead to dysregulated TGF-β signaling, which may enhance SMAD2/3 phosphorylation (61). This results in conversion of a tolerogenic pathway to a pro-allergic one by producing dysfunctional Tregs and by enhancing transcription of IL-9 and other pro-allergic mediators (62).
Key defects in intracellular signaling components leading to altered cytokine signaling defects
4
JAK1 GOF
4.1
Germline JAK1 GOF mutations lead to constitutive activation of the Janus kinase 1 protein, leading to immune dysregulation due to unchecked STAT phosphorylation (63). Mechanistically, JAK1 hyperactivity skews CD4^+^ T-cell differentiation toward a Th2 phenotype while suppressing Th1 responses (63). This results in amplification of allergic inflammation (63). These variants lead to novel monogenic immune dysregulation syndrome, termed JAACD**—**JAK1-associated Autoimmunity, Atopy, Colitis, and Dermatitis. Affected individuals exhibit a syndromic phenotype characterized by early-onset atopic disease, autoimmune features, severe dermatitis, and inflammatory bowel manifestations such as colitis (64). Del Bel et al. described the first germline GOF mutation in humans in JAK1, resulting in an alanine to aspartate substitution at position 634 (65). Three more variants were identified in JAK1, namely, S703I, H596D, and C787F, from patients with a similar clinical phenotype (66–68). Recently, Horesh et al. described 59 patients with JAACD spectrum harbouring four JAK1-GOF variants (p.E139K, p.R506C, p.S700N, and p.V985I). These patients share a common phenotype of severe atopy with autoimmunity and immune dysregulation (64).
ERBB2IP (ERBIN) LOF
4.2
ERBB2IP encodes ERBIN, a scaffold protein that links activated STAT3 to SMAD2/3 complexes, sequestering them in the cytoplasm and thereby restricting TGF-β signaling (49, 69). Impaired STAT3 signaling can decrease ERBIN levels resulting in disrupted regulation of TGF-β and consequent increase in Tregs. Autosomal-recessive LOF variants in ERBB2IP disrupt this regulatory axis, leading to excessive SMAD2/3 nuclear translocation, enhanced Treg proliferation, and paradoxical Th2 polarization (49). Clinically, ERBIN-deficient patients exhibit anomalies reminiscent of STAT3-HIES, despite a distinct molecular etiology. Patients may present with severe atopic dermatitis, eosinophilic gastrointestinal diseases, elevated IgE, and connective-tissue disorders. Emerging biologic therapy, such as IL-4Rα blockade with dupilumab has shown promise in reducing Th2-driven inflammation in this disorder (70).
Future perspectives
5
Next-generation sequencing (NGS) is revolutionizing the diagnosis of primary atopic disorders (PADs) by enabling rapid identification of disease-causing variants. This technology is especially valuable in patients with complex or treatment-refractory presentations, where it can resolve long-standing diagnostic challenges. However, as the use of NGS expands, a growing number of variants of uncertain significance (VUS) or cases lacking identifiable pathogenic variants are being reported. These findings often complicate clinical decision-making and may necessitate extensive additional testing.
Large-scale analyses of genetic testing across hereditary diseases have underscored the growing burden of variants of uncertain significance (VUS). In a study involving over 1.6 million individuals, 41% had at least one VUS, and nearly one-third received only VUS results (71). Despite efforts, only 7% of unique VUSs were reclassified as pathogenic or likely pathogenic, often taking over two years (71). These findings underscore the broader systemic challenge posed by VUSs across rare disease diagnostics and highlight the need for structured interpretive frameworks that could also benefit the PAD diagnostic landscape.
Looking forward, decision-making around functional validation of variants must be guided by integrated criteria, including in silico prediction tools, family segregation analysis, phenotypic concordance, and population frequency data (72). While functional assays remain the gold standard for confirming pathogenicity, they are resource-intensive and often inaccessible in routine clinical settings.
Future diagnostic frameworks must prioritise variants with the highest clinical relevance. Innovative tools such as multiplexed assays of variant effect (MAVEs) offer a promising, high-throughput approach to functional validation (73). Concurrently, emerging efforts to harmonise the interpretation of single-nucleotide variants (SNVs) and copy-number variants (CNVs) are streamlining variant classification in rare diseases (74).
To fully realise the promise of precision medicine in PADs, future efforts should focus on building standardised, scalable, and integrative diagnostic pipelines. Such systems will be essential for accelerating diagnosis, guiding targeted therapies, and ultimately improving clinical outcomes for patients.
Conclusion
6
PADs represent a critical intersection between monogenic immune dysregulation and severe allergic inflammation. Cytokine signaling defects involving JAK-STAT and TGF-β pathways can result in profound allergic phenotypes often misdiagnosed as common atopy. Timely diagnosis of the pathogenetic defect using advanced next-generation sequencing is essential to deliver targeted, immune-based therapies. As our understanding of PADs continues to expand, personalized approach will eventually be the standard of care for affected individuals.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Notarangelo LD Bacchetta R Casanova JL Su HC. Human inborn errors of immunity: an expanding universe. Sci Immunol. (2020) 5(49):eabb 1662. 10.1126/sciimmunol.abb 166232651211 PMC 7647049 · doi ↗ · pubmed ↗
- 2Tangye SG Al-Herz W Bousfiha A Chatila T Cunningham-Rundles C Etzioni A Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2020) 40(1):24–64. 10.1007/s 10875-019-00737-x 31953710 PMC 7082301 · doi ↗ · pubmed ↗
- 3Amaya-Uribe L Rojas M Azizi G Anaya J-M Gershwin ME. Primary immunodeficiency and autoimmunity: a comprehensive review. J Autoimmun. (2019) 99:52–72. 10.1016/j.jaut.2019.01.01130795880 · doi ↗ · pubmed ↗
- 4Lyons JJ Milner JD. Primary atopic disorders. J Exp Med. (2018) 215(4):1009–22. 10.1084/jem.2017230629549114 PMC 5881472 · doi ↗ · pubmed ↗
- 5Palmer CN Irvine AD Terron-Kwiatkowski A Zhao Y Liao H Lee SP Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. (2006) 38(4):441–6. 10.1038/ng 176716550169 · doi ↗ · pubmed ↗
- 6Bitoun E Micheloni A Lamant L Bonnart C Tartaglia-Polcini A Cobbold C LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in netherton syndrome. Hum Mol Genet. (2003) 12(19):2417–30. 10.1093/hmg/ddg 24712915442 · doi ↗ · pubmed ↗
- 7Gaddameedhi S Selby CP Kemp MG Ye R Sancar A. The circadian clock controls sunburn apoptosis and erythema in mouse skin. J Invest Dermatol. (2015) 135(4):1119–27. 10.1038/jid.2014.50825431853 PMC 4366313 · doi ↗ · pubmed ↗
- 8Castagnoli R Lougaris V Giardino G Volpi S Leonardi L La Torre F Inborn errors of immunity with atopic phenotypes: a practical guide for allergists. World Allergy Organ J. (2021) 14(2):100513. 10.1016/j.waojou.2021.10051333717395 PMC 7907539 · doi ↗ · pubmed ↗