Understanding the microbiome in autologous haemopoietic stem cell transplant for multiple sclerosis
Jun Yin, Nadeem O. Kaakoush, Jennifer Massey, Mark Danta

TL;DR
This study explores how autologous stem cell transplant affects the gut and oral microbiome in people with multiple sclerosis, revealing distinct microbial changes compared to those on Natalizumab.
Contribution
This is the first study to investigate the impact of AHSCT on the microbiome in multiple sclerosis patients.
Findings
AHSCT cohort had lower oral species richness compared to the Natalizumab group.
Significant differences in oral beta diversity were observed between the two cohorts.
AHSCT subjects showed increased Porphyromonas and decreased Veillonella in oral taxa.
Abstract
MS is a chronic inflammatory and degenerative disease of the central nervous system (CNS) resulting in neurological deficits associated with physical and/or cognitive disability. The gut microbiome can interact with the CNS and immune system through various molecular pathways and has been previously implicated in MS. Autologous Haematopoietic Stem Cell Transplant (AHSCT) in MS arrests inflammatory disease and has evidence of long-term therapeutic benefit. To date, no study has explored the effect of AHSCT on the gut microbiome in people with MS. The microbiome of people with MS (pwMS) undergoing AHSCT was compared with pwMS on Natalizumab (NTZ). Longitudinal microbiome analysis was also conducted within the AHSCT cohort at two timepoints. Amplicon sequencing of the 16S ribosomal RNA V3–4 region (Illumina MiSeq) was used to evaluate alpha and beta diversity, oral-stool microbiota…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
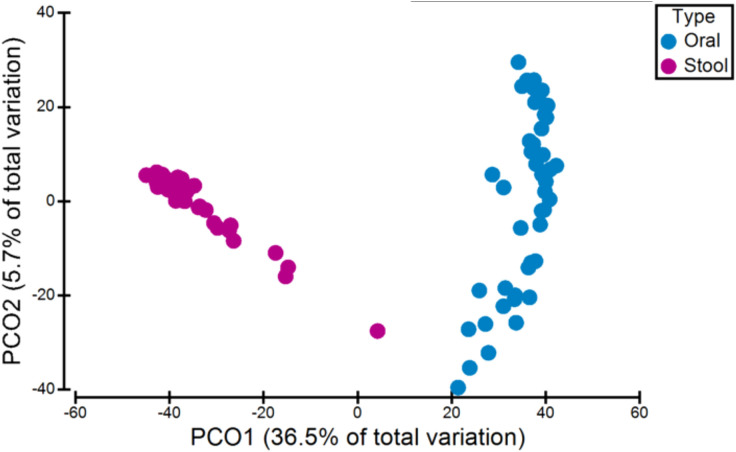 Figure 1
Figure 1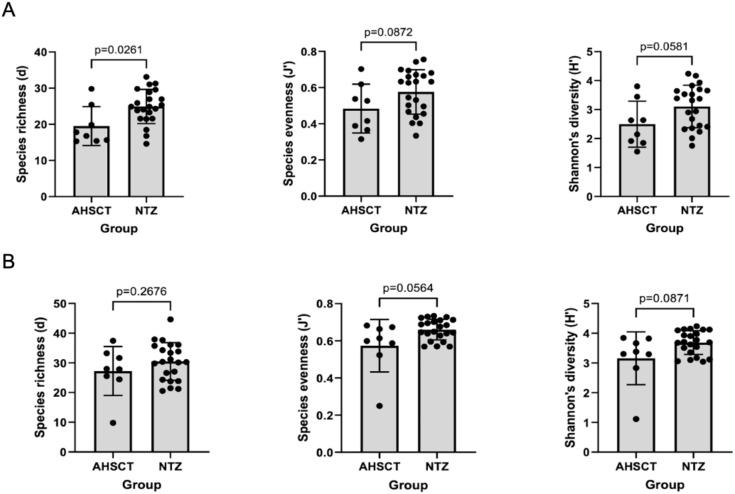 Figure 2
Figure 2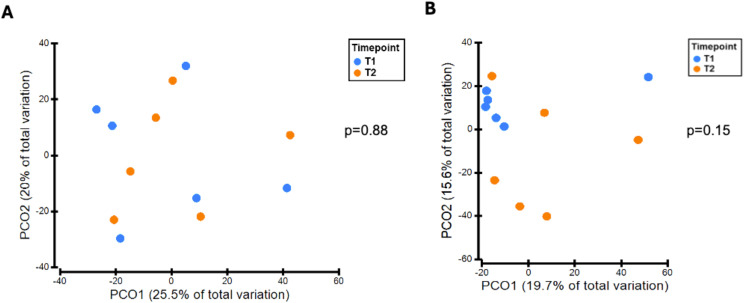 Figure 3
Figure 3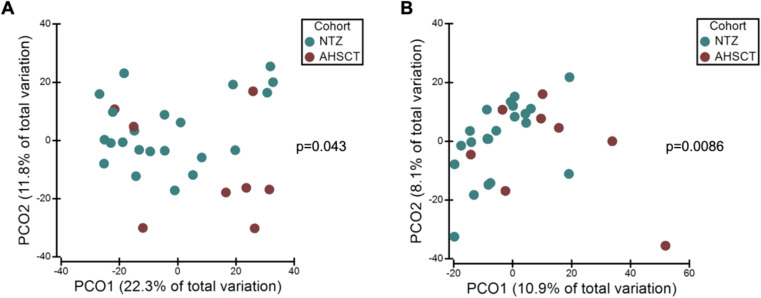 Figure 4
Figure 4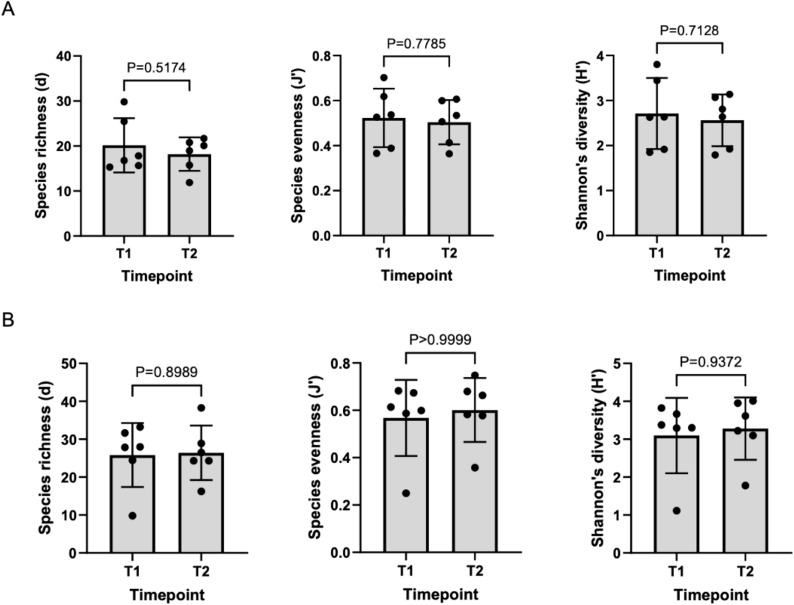 Figure 5
Figure 5| Inclusion Criteria |
|---|
| • Diagnosis of relapsing MS made by a neurologist according to the 2017 revised McDonald’s criteria ( |
| Exclusion Criteria |
| • Any patient on the study treatment arm deemed not suitable for transplant by a consensus of a HSCT multidisciplinary team composed of specialist doctors. |
| Natalizumab Patients (n = 22) | AHSCT Patients (n = 8) | |
|---|---|---|
| Age (years) | ||
| Median ± SD | 36.5 ± 10.8 | 37.5 ± 6.7 |
| Sex, n (%) | ||
| Male | 2 (9.1) | 2 (25.0) |
| Female | 20 (90.9) | 6 (75.0) |
| Ethnicity (%) | ||
| Anglo-Celtic | 15 (68.2) | 7 (87.5) |
| European | 4 (18.2) | 0 (0) |
| East/South-East Asian | 1 (4.5) | 0 (0) |
| Aboriginal | 0 (0) | 1 (12.5) |
| Middle Eastern | 2 (9.1) | 0 (0) |
| Type of MS (%) | ||
| Relapsing Remitting | 21 (95.5) | 8 (100) |
| Secondary Progressive | 1 (4.5) | 0 (0) |
| EDSS | ||
| Median ± SD | 2.0 ± 0.9 | 4.8 ± 1.7 |
| Smoking, n (%) | ||
| Smoker | 0 (0) | 0 (0) |
| Non-smoker | 22 (100) | 8 (100) |
| Alcohol, n (%) | ||
| >3 standard drinks per week | 5 (22.7) | 0 (0) |
| <3 standard drinks per week | 17 (77.3) | 8 (100) |
| Antibiotics used in last 12 months, n (%) | ||
| Yes | 6 (27.3) | 7 (87.5)* |
| No | 16 (72.7) | 1 (12.5) |
| Patient Number | Timepoint 1 (Stool and Oral) Sample | Timepoint 2 (Stool and Oral) Sample |
|---|---|---|
| Patient 1 | Baseline (pre-transplant) | 7 months |
| Patient 2 | Baseline (pre-transplant) | 7 months |
| Patient 3 | Baseline (pre-transplant) | 12 months |
| Patient 4 | Baseline (pre-transplant) | 12 months |
| Patient 5 | Baseline (pre-transplant) | 10 months |
| Patient 6 | Baseline (pre-transplant) | 10 months |
|
| |||
|---|---|---|---|
| OTUs | q-value corrected for FDR | Fold Change AHSCT/NTZ | Bacterial species |
| Porphyromonas_OTU69 | 0.015 | 1.16 | Gram-negative anaerobe associated with numerous systemic inflammatory and neurodegenerative disease ( |
| Veillonella_OTU2 | 0.100 | 0.27 | Gram-negative obligate anaerobe ( |
|
| |||
| Lachnospiraceae_unclassified_OTU406 | 0.032 | 3.39 | Gram-positive anaerobe associated with SCFA production ( |
| Enterocloster_OTU121 | 0.056 | 5.37 | Gram-positive obligate anaerobe associated with SCFA production ( |
| Roseburia_OTU68 | 0.089 | 0.16 | Gram-positive obligate anaerobe associated with SCFA production ( |
| Bifidobacterium_OTU405 | 0.089 | 2.10 | Gram-positive, facultative anaerobe associated with SCFA production ( |
| Parabacteroides_OTU15 | 0.091 | 1.75 | Gram-negative anaerobe involved in SCFA production ( |
| Hominimerdicola_OTU25 | 0.091 | 0.20 | Gram-positive obligate anaerobe. |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGut microbiota and health · Oral health in cancer treatment · Salivary Gland Disorders and Functions
Introduction
1
Multiple sclerosis (MS) is a chronic, immune-mediated disease of the central nervous system (CNS) which affects an estimated 2.8 million people worldwide (1). With the average age of diagnosis ranging from 30–40 years old (2), MS causes significant physical and cognitive disability (3).
Genes within the human leukocyte antigen (HLA) complex remain the strongest genetic risk factor for MS (4). However, other epidemiologic risk factors include exposure to Epstein Barr virus, vitamin D levels, geographical location such as latitude, obesity in adolescence and smoking (5). Intestinal microbiota has also emerged as a potential factor influencing MS pathogenesis (6). Activation of an abnormal adaptive immune response is central to disease initiation (7). Self-reactive T and B lymphocytes infiltrate the CNS, crossing the blood-brain barrier (BBB) and triggering inflammatory cascades that result in glial and neuronal injury (8).
Natalizumab (NTZ), is a monoclonal antibody which inhibits T-cell migration into the CNS via α4-integrin inhibition (9). However, its action is non-specific, also disrupting T-cell circulation within the gut (10). This makes NTZ an intriguing drug for studying the gut-immune axis and its potential alternate pathways of action in MS. Autologous hematopoietic stem cell transplant (AHSCT) has demonstrated the potential for immune reconstitution, particularly in patients with more severe, DMT-refractory MS (11). AHSCT involves high-dose chemotherapy to eradicate autoreactive immune cells, followed by the infusion of autologous stem cells to rebuild a novel immune system (7).
The gut microbiome is increasingly recognized as a key player in modulating systemic immunity and CNS inflammation. Microbial metabolites, such as short-chain fatty acids (SCFAs) and neurotransmitters, influence peripheral immune function, while cytokines and chemokines derived from gut bacteria can disrupt CNS homeostasis (12). Conversely, CNS-derived biochemical changes can alter gut microbial composition through the hypothalamic-pituitary-adrenal (HPA) axis (13). Specific gut bacteria, such as segmented filamentous bacteria (SFB), are known to drive T-helper 17 cell differentiation, which is implicated in autoimmune diseases, including MS (14). Dysbiosis, an imbalance in microbial composition or metabolites, can exacerbate neuroinflammation by disrupting intestinal and systemic immune homeostasis (15). Although numerous studies have identified differences in stool microbial taxa between people with MS (pwMS) and healthy controls (HC) (16–21), these findings are largely correlative. It remains unclear whether dysbiosis contributes to MS pathogenesis or is secondary to the disease and its treatments. There are also limited studies characterising the oral microbiome in pwMS. Furthermore, while research on gut microbiota’s role in immune reconstitution following hematopoietic stem cell transplantation (HSCT) in haematological malignancies has revealed intriguing associations, similar studies in pwMS have yet to be conducted.
This present study aims to firstly characterise the differences in the oral and stool microbiome of people with MS in remission on treatment (NTZ) compared with those pre-AHSCT, refractory to available DMT. Secondly, for pwMS undergoing AHSCT, analysis of longitudinal changes in oral and stool microbiota could help identify microbial shifts associated with disease activity or treatment effects, and explore their potential relevance to immune reconstitution.
Method
2
Study outline
2.1
An observational cohort study was conducted by comparing the oral and stool microbiome of pre-AHSCT pwMS with a control group of pwMS undergoing treatment with NTZ (St. Vincent’s Hospital, Sydney HREC 2021/ETH11173). This allowed for a comparison of the microbiome in active DMT-refractory MS and stable, treated MS. Within the AHSCT cohort, a longitudinal analysis of the oral and stool microbiome was then conducted throughout treatment. All patients provided written consent prior to sample collection.
Cohorts
2.2
NTZ Control Cohort: Patients, aged 18-65, with MS as determined by a neurologist according to the 2017 revised McDonald’s criteria (22) were suitable for inclusion in the control cohort. Recruited patients had been receiving NTZ for 3 or more months, 300mg of intravenous NTZ every 4–6 weeks. Pre- and post-AHSCT samples were collected from these patients.
AHSCT Cohort: All patients undergoing AHSCT as part of the AHSCT in Multiple Sclerosis (AIMS) study were required to contribute stool and saliva samples as part of the interventional clinical trial. Participants were off immunotherapy at clinically appropriate wash-out periods prior to the collection of baseline, pre-ASHCT samples (Appendix 1). This allowed for a snapshot of the gut microbiome in ‘active’, DMT-refractory MS. See Table 1. for the inclusion and exclusion criteria of this cohort.
Patient recruitment
2.3
Patients were recruited between 1 Jan 2022 and 1 July 2024. Each patient was assigned a unique study ID, with relevant clinical and demographic data recorded in the institutional REDCap database.
Sample collection and storage
2.4
INVITEK Molecular kits were used to collect faecal samples and DNAGenotek kits were used to collect oral samples. For AHSCT patients, sample collection was scheduled before AHSCT treatment (baseline samples), and 6 months post-AHSCT. AHSCT participants were provided with self-collection kits at the 6-month post-AHSCT appointment. However, as samples were self-collected at home and returned via post, actual collection times ranged from 7 to 12 months. Samples were aliquoted and stored at -80°C, which is regarded as the gold standard (23).
Nucleic acid extraction
2.5
DNA extraction was conducted at the Microbiome Research Centre, Kogarah, NSW. Frozen stool samples were processed using the INVITEK™ PSP DNA extraction kit. Frozen oral samples were processed using the Qiagen™ DSP pathogen DNA extraction kit.
Sequencing and data analysis
2.6
The 16S rRNA V4 regions were amplified by PCR and sequenced on the MiSeq platform (Illumina) using the 2×300 bp paired-end protocol. Sequencing was performed at the UNSW Ramaciotti Centre for Genomics. Raw sequencing data was analysed using Mothur v1.48.1 and vSearch v2.22.1 on the UNSW Katana server (24). The Ribosomal Database Project (RDP) v19 was used as the reference database for taxonomic classification. Operational taxonomic units (OTUs) were used to group clusters of uncultivable microorganisms with sequence similarity as an analytical unit. The numbers represented the degree of importance; the lower the number the more prevalent the genus (i.e. OTU05 is more prevalent than OTU26). Similarity was detected by the 16S taxonomic marker gene and was used to classify microbial individuals at different taxonomic levels.
Primer-E v6 was used to calculate alpha diversity measures including Margalef’s species richness, Pielou’s evenness and Shannon’s diversity index. For both stool and oral samples, the Shaprio-Wilk test was used to test for normality of data. Alpha diversity of normally distributed data was analysed with unpaired t-tests while data for which didn’t fit a normal distribution, Mann-Whitney U tests were used. Beta diversity was calculated with Bray-Curtis dissimilarities from OUT relative abundances (%) that were transformed by square root. Principal-coordinate analysis (PCoA) and permutational multivariate analysis of variance (PERMANOVA) was then calculated. The comparison between the oral and stool microbiota was performed with a PCoA and PERMANOVA on a Bray-Curtis resemblance matrix, and the distance relationship between the oral and stool microbiota was calculated with a Mann-Whitney U test. GraphPad Prism v9.4 was used for statistical analysis.
For taxonomic classification, per taxon analysis was performed using ZicoSeq (25). Counts were log-transformed and only taxa that contributed to at least 1% to the oral and stool microbiota were included. An unpaired t-test was applied and p-values were corrected for false discovery rates (FDR) using the Benjamini- Hochberg method (BH) to form a q-value.
Results
3
Subject characteristics
3.1
A total of 30 participants with MS were enrolled into the study: 22 in the NTZ cohort and 8 in the AHSCT cohort. The two cohorts were well-matched in terms of sex and age. The AHSCT cohort had a higher Expanded Disability Status Scale (EDSS) median score and higher 12-month antibiotic exposure. No patients in the AHSCT cohort received NTZ within 6-months of treatment. Demographic information about the cohorts is summarised in Table 2. Baseline oral and stool samples were compared from all patients in the AHSCT and the NTZ control cohorts. Of the 8 AHSCT patients who provided baseline oral and stool samples (T1 samples), 6 provided longitudinal samples at a second timepoint (T2 samples) with a median time of 10-months (interquartile range: 5-months) (see Table 3).
Comparison of baseline oral and stool microbiota
3.2
To analyse the overall interindividual difference between stool and oral samples (beta diversity), PCoA and a non-parametric multivariate statistical permutation test (PERMANOVA) was performed. There was a highly significant difference between oral and stool microbiota (p=0.0001, PERMANOVA) (see Figure 1).
Variation in microbiota (beta-diversity) between all oral and stool samples. There was a significant difference between oral and stool microbiota (p=0.0001, PERMANOVA).
AHSCT vs NTZ cohorts
3.3
The AHSCT cohort had significantly lower species richness of oral microbiota compared to the NTZ cohort (p=0.026) (Figure 2A). While there was no significant difference in evenness (p=0.0872) and diversity (p=0.0581) between the two cohorts, there was a trend to lower evenness and diversity in the AHSCT group. While generally lower in the AHSCT cohort, there was no significant difference in the alpha diversity of the stool microbiota richness (p=0.2676), evenness (p=0.0564) and diversity (p=0.0871) between the AHSCT and NTZ cohorts (Figure 2B). There was a significant difference in the oral beta diversity (p=0.043, PERMANOVA) (Figure 3A) and stool beta diversity (p=0.0086, PERMANOVA) (Figure 3B) between the AHSCT and NTZ groups.
(A) Comparison of oral alpha diversity between AHSCT and NTZ cohort; (B) Comparison of stool alpha diversity between AHSCT and NTZ cohorts. Normality of datasets was determined using Shapiro-Wilk tests. Unpaired t-tests (parametric) or Mann-Whitney U tests (non-parametric) were then used to compare alpha diversity statistics, including Margalef’s richness, Pielou’s evenness and Shannon’s diversity. For the oral samples, there was a trend towards lower alpha diversity in the AHSCT cohorts. However, only species richness was statistically significant (p=0.0261).
(A) Oral microbiota variation between AHSCT and NTZ cohorts, (B) Stool microbiota variation between AHSCT and NTZ cohorts. Beta diversity statistics were generated using PCoA and PERMANOVA. PCoA was performed on Bray Curtis resemblance matrix of square root transformed relative abundances. There were significant differences in beta-diversity for oral (p=0.043, PERMANOVA) and stool (p=0.0086, PERMANOVA) samples between AHSCT and NTZ cohorts.
Longitudinal AHSCT analysis
3.4
There was no significant difference in alpha diversity between T1 and T2 oral microbiota samples (Figure 4A). T1 and T2 stool microbiota samples also revealed no significant differences in alpha diversity (Figure 4B). Comparison of oral samples at T1 and T2 showed no differences in beta diversity (p=0.88, PERMANOVA) (Figure 5A). Similarly, comparison of stool samples at T1 and T2 showed no differences in beta diversity (p=0.15, PERMANOVA) (Figure 5B).
(A) Comparison of oral alpha diversity between T1 and T2 samples, (B) Comparison of stool alpha diversity between T1 and T2 samples. The differences in alpha diversity, including Margalef’s richness, Pielou’s evenness and Shannon’s diversity, between T1 and T2 samples was determined using unpaired t-tests and Mann Whitney U tests. Analysis of both oral and stool microbiota revealed no differences in alpha diversity between T1 and T2 samples.
(A) Variation in oral microbiota between T1 and T2 samples, (B) Variation in stool microbiota between T1 and T2 samples. There was no significant difference in oral (p=0.88, PERMANOVA) or stool (p=0.15, PERMANOVA) beta-diversity between T1 and T2 samples.
Taxa analysis of NTZ and AHSCT cohorts at baseline
3.5
A per taxa analysis was performed to identify operational taxonomy units (OTUs) which contributed to differences in beta diversity between AHSCT and NTZ cohorts. The taxa were identified at the genus level, their lowest possible level of classification (Table 4). For stool samples, the AHSCT group had greater relative abundances of an unclassified genus of Lachnospiraceae, Enterocloster, Bifidobacterium and Parabacteroides and lower abundances of Roseburia and Hominimerdicola. For oral microbiota, the AHSCT group has greater relative abundances of Porphyromonas and decreased abundances of Veillonella.
Discussion
4
Comparison of AHSCT and NTZ cohorts
4.1
Despite emerging evidence linking oral bacteria to various systemic diseases (30), the role of oral microbiota in MS remains underexplored. While previous research predominantly compares individuals with MS to healthy controls (HC) (16–21), this study offers a novel approach by examining microbiome differences across MS patients with differing treatment exposures and disease activity levels. Specifically, we focused on patients undergoing AHSCT, who typically have active, treatment-refractory MS, and compared them to patients receiving NTZ, with controlled, non-active MS.
A key finding in this study was the lower species richness of oral microbiota in the ASHCT group compared to the NTZ group. The correlation of reduced alpha diversity in the more active AHSCT cohort raises the possibility that increased oral diversity may have a protective effect in MS. This supports the observations of a longitudinal study comparing the microbiome of pwMS undergoing ocrelizumab therapy. The study reported decreased oral alpha diversity in MS patients compared to HC, revealing that diversity patterns were most pronounced in patients with higher disease activity and disability (31). It is unclear whether these oral microbial changes are an effect of MS therapy or indirectly modulate MS disease activity and this requires further investigation.
Our findings of increased oral Porphyromonas in the AHSCT group is also notable, given previous studies have reported an increased abundance of oral Porphyromonas in pwMS compared to HC (32, 33). Additionally, Porphyromonas gingivalis has been associated with several systemic diseases, including Alzheimer’s disease, various cancers, atherosclerosis, periodontitis, and systemic inflammation (34–36). Its pathogenic potential can be attributed to its lipopolysaccharides, particularly the lipid A structure, which can trigger proinflammatory responses by activating the NF-κB signalling pathways and promoting the secretion of inflammatory cytokines (37). In EAE, a murine model of demyelination, when immune-competent glial cells of the CNS are exposed to antigens of Porphyromonas gingivalis, immune responses and disability scores are exacerbated. However, the significance of this finding needs to be confirmed in human studies (38, 39).
We observed a significant depletion of oral Veillonella in ‘active’ MS (pre-AHSCT cohort) compared to treated MS (NTZ group). A recent study comparing pwMS to HC found increased abundance of oral Veillonella in MS populations. Several factors may explain these divergent findings. Veillonella is part of the core oral microbiome in healthy individuals and its abundance can be influenced by confounders such as periodontal disease, oral hygiene, and dietary habits (40, 41). Notably, a greater proportion of AHSCT patients had received antibiotics in the 12 months prior to sample collection, which may have contributed to the observed depletion, as systemic antibiotic use is known to disrupt oral microbial communities even when not directly targeting the oral cavity (42). It is also important to consider that our comparison involves two MS subgroups with differing disease activity and treatment exposure, rather than a comparison with healthy controls. As such, the observed differences in Veillonella abundance may reflect treatment effects, immunological state, or other systemic influences. Thus, further studies with larger MS cohorts with controlled oral confounders are required to are required to clarify Veillonella’s role and clinical relevance in the context of MS.
Our study revealed no differences in stool alpha diversity between AHSCT and NTZ groups. Similar results were observed in the iMSMS study (21) which found no differences in alpha diversity between treated and non-treated MS populations. Beta diversity between the AHSCT and NTZ cohorts, however, was significantly different, highlighting the possibility that varying MS disease states may be associated with stool microbial composition.
Specific taxa findings revealed increased relative abundances of an unclassified genus of Lachnospiraceae, Enterocloster, Bifidobacterium and Parabacteroides in the AHSCT group compared to the NTZ group. While the lack of HCs in this study makes it difficult to confirm whether these bacteria are significantly altered in MS populations compared to healthy individuals, evidence from numerous studies show they are likely to be implicated in MS (18, 20, 21, 43–45). Consistent with previous studies, we found lower abundances of Roseburia and Hominimerdicola in the AHSCT group. Roseburia has been noted to decrease in MS patients compared to healthy controls (45, 46), suggesting a protective role in MS. It is hypothesised that Roseburia may ameliorate MS through its production of the butyrate, an SCFA with anti-inflammatory properties (47). It should be acknowledged that prior work has conversely suggested that Lachnospiraceae, Bifidobacteirum and Parabacteroides have decreased abundance in untreated MS populations compared to HC (43–45). Specifically, Parabacteroides distasonis has been shown to induce regulatory T cells that produce anti-inflammatory IL-10 (48). Acetate, a SCFA highly produced by Bifidobacteirum, regulates intestinal inflammation by stimulating the GPR43 receptor which in turn, inhibits the secretion of pro-inflammatory IL-18 (49). These conflicting results highlight the complex relationship between specific bacterial taxa, MS and the immune system which can be modulated by other factors, such as disease stage and treatment status.
Interestingly, a consistent pattern emerging from both our study and previous research is the association of MS with SCFA-producing bacteria (46). Five out of the six taxa identified in our study responsible for the stool beta-diversity differences are involved in the production of SCFAs. SCFAs, particularly butyrate and acetate, can promote anti-inflammatory responses by supporting regulatory T cells but also influence pro-inflammatory Th1 and Th17 cell differentiation in certain contexts (50, 51). Increased acetate levels in MS patients correlated with higher disability levels measured by their EDSS (52). While our findings highlight a strong association between stool microbiota and their metabolites with MS, further investigation into bacterial metabolites like SCFAs in MS populations is required to elucidate the exact mechanisms involved.
A potential confounding factor is the effect of NTZ on both oral and gut microbiota. Currently, there is limited evidence on the impact of NTZ on oral microbial composition. However, research has demonstrated that NTZ treatment can reduce specific stool bacterial populations, including some that produce short-chain fatty acids (SCFAs), such as Bacteroides and Bifidobacterium species, which are key to gastrointestinal health (21, 53). While it is believed NTZ’s mechanism of action against MS involves the inhibition of T-cell trafficking to the CNS, it also impacts T-cell circulation within the GIT (54). This immunomodulatory action on the gut could influence microbial composition, possibly representing an alternative pathway by which this therapy acts against MS. NTZ-treated patients were chosen as the comparator group due to their clinical stability, consistent treatment duration, and lack of recent immunosuppressive induction, which make them a valuable reference for understanding microbiome profiles in MS under controlled treatment conditions. However, the differences in microbial composition between the AHSCT and NTZ cohorts may be due to treatment-specific effects, in addition to or instead of disease activity.
Longitudinal analysis
4.2
Analysis of T1 (pre-transplant) and T2 (post-transplant) samples for both oral and stool demonstrated no significant differences in alpha- or beta-diversity, revealing that the diversity of stool and oral microbial populations remained relatively stable post-AHSCT, despite the peri-transplant use of chemotherapy, steroids and antimicrobials. Our findings contrast numerous studies where a marked reduction in stool alpha diversity post-allogenic HSCT was observed (55, 56). In Khan et al., the largest and first multicentre study of stool microbiota composition in autologous HSCT, alpha diversity of post-transplant stool samples was also significantly decreased (57). A plausible explanation for the discrepancy in our results lies in the different post-transplant timeframes assessed across studies. While the aforementioned studies collected post-transplant stool samples within the at 0–100 days post-transplant, our study examined samples collected between 7–12 months post-transplant. This extended period may have allowed the stool microbial population to recover and return to pre-transplant diversity levels, and this return to ‘baseline’ is a fascinating observation in itself. This is supported by longitudinal data from Khan et al., where microbial diversity in stool samples reached its lowest point at 14–17 days post-transplant but subsequently increased (57).
The diversity of gut microbiome before and after transplant has also been linked to improved clinical outcomes. Notably, research has indicated that higher gut microbiota diversity pre-transplant correlates with improved overall survival and a lower incidence of acute graft-versus-host disease (GVHD) in allogeneic HSCT patients (58–60). Additionally, lower diversity during the peri-engraftment phase has been associated with worse outcomes in autologous HSCT patients (57). While these correlations have primarily been drawn from malignant-related HSCT cases, they suggest that the stability of the gut microbiome post-AHSCT in our cohort of MS patients may be linked to positive clinical outcomes, especially since all patients our AHSCT cohort have met the “no evidence of disease activity” (NEDA) criteria and have not progressed in terms of MS disability. However, a larger sample size and correlation to long-term follow up data is required to validate such relationships.
Limitations and future direction
4.3
This study was limited by the relatively small sample size of 30 patients. In particular, recruitment for the AHSCT cohort was restricted by the number of MS patients who met the inclusion criteria for the AiMS study at St Vincent’s Hospital, and ongoing challenges through 2022 with COVID-19. On average, only 5–10 patients undergo AHSCT for MS each year, which has limited the pace of recruitment. As this pilot study is ongoing, we anticipate that a more comprehensive analysis of longitudinal changes in the oral and gut microbiome will be possible as additional data are collected across multiple timepoints. These future analyses may enable correlation of microbial profiles with clinical outcomes, including MS severity and markers of immune reconstitution following AHSCT.
Although the NTZ and AHSCT cohorts were broadly matched for age, sex, and ethnicity, minimising the likelihood of major demographic confounding, statistical analyses to explore associations between individual demographic variables and microbiota composition were not conducted. This was due to the limited sample size and lack of statistical power for reliable subgroup analysis but can also be addressed as additional data is collected.
Due to small numbers, confounding factors in each cohort such as gender, diet or recent antibiotics couldn’t be accounted for and may influence GIT microbial composition. Oral health parameters such as periodontal status and oral hygiene practices were not assessed. Given the known influence of these factors on oral microbial composition, their omission may have impacted our findings (61–63). In future studies, adopting a household paired system whereby the patient with MS and controls live in the same household could address this issue as both individuals would have similar diets and lifestyle factors. Incorporating standardised oral health assessments may also better control for oral microbiome variability.
The variability in timing of post-AHSCT sample collection (between 7–12 months) is also acknowledged as a limitation. This arose due to the logistics of home sampling and postal return. While all samples were collected after the acute transplant phase, this range may have influenced microbiota composition. As more data are collected, we intend to stratify post-AHSCT samples into narrower timepoints (e.g. 6–8 months vs 9–12 months) to examine trends with greater granularity.
Additionally, Natalizumab’s effects on immune cell trafficking in the gastrointestinal tract may independently influence microbiota composition. This introduces a potential confounder when interpreting differences between cohorts and highlights the need for future studies incorporating additional control groups.
This study did not aim to compare MS cohorts with a healthy control population, as this has been extensively previously explored. However, the lack of an internal control is another limitation in our study. Consequently, it was questionable whether the differences between our two cohorts were due to the differing treatments or the varying severity of disease. However, this limitation was partially overcome by supporting evidence from studies which found that the specific bacterial taxa identified in our study were previously implicated in MS.
Conclusion
5
This pilot study identified specific microbiome changes, particularly in the oral alpha diversity and abundance of specific bacteria which may play roles in MS progression or pathogenesis. Furthermore, longitudinal analysis combined with clinical follow-up data alludes to a potential relationship between stool microbial diversity and positive AHSCT outcomes. Ongoing recruitment and further analysis will determine the validity of these findings and may provide a novel therapeutic target in MS treatment.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Walton C King R Rechtman L Kaye W Leray E Marrie RA. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult Scler. (2020) 26:1816–21. doi: 10.1177/1352458520970841 PMC 772035533174475 · doi ↗ · pubmed ↗
- 2Habbestad A Willumsen JS Aarseth JH Grytten N Midgard R Wergeland S. Increasing age of multiple sclerosis onset from 1920 to 2022: a population-based study. J Neurol. (2024) 271:1610–7. doi: 10.1007/s 00415-023-12047-9 PMC 1097305038097800 · doi ↗ · pubmed ↗
- 3Ford H. Clinical presentation and diagnosis of multiple sclerosis. Clin Med (Lond). (2020) 20:380–3. doi: 10.7861/clinmed.2020-0292 PMC 738579732675142 · doi ↗ · pubmed ↗
- 4Moutsianas L Jostins L Beecham AH Dilthey AT Xifara DK Ban M. Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat Genet. (2015) 47:1107–13. doi: 10.1038/ng.3395 PMC 487424526343388 · doi ↗ · pubmed ↗
- 5Hollenbach JA Oksenberg JR. The immunogenetics of multiple sclerosis: A comprehensive review. J Autoimmun. (2015) 64:13–25. doi: 10.1016/j.jaut.2015.06.010 26142251 PMC 4687745 · doi ↗ · pubmed ↗
- 6Berer K Krishnamoorthy G. Microbial view of central nervous system autoimmunity. FEBS Lett. (2014) 588:4207–13. doi: 10.1016/j.febslet.2014.04.007 24746689 · doi ↗ · pubmed ↗
- 7Massey JC Sutton IJ Ma DDF Moore JJ. Regenerating immunotolerance in multiple sclerosis with autologous hematopoietic stem cell transplant. Front Immunol. (2018) 9:410. doi: 10.3389/fimmu.2018.00410 29593711 PMC 5857574 · doi ↗ · pubmed ↗
- 8Voet S Prinz Mvan Loo G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol Med. (2019) 25:112–23. doi: 10.1016/j.molmed.2018.11.005 30578090 · doi ↗ · pubmed ↗