Concomitant Systemic Autoinflammatory Diseases: Diagnostic and therapeutic challenges
Eman Al Masroori, Mahadev J. Mal, Reem Abdwani

TL;DR
A 3-month-old infant had overlapping symptoms of two rare inflammatory diseases, requiring advanced genetic testing and combined treatment for effective management.
Contribution
Highlights the diagnostic and therapeutic challenges of coexisting systemic autoinflammatory diseases and the role of somatic mosaicism.
Findings
The infant had NOMID confirmed by NLRP3 somatic mosaicism despite initial negative genetic tests.
Coexisting FMF was identified through MEFV gene mutations, requiring combined treatment with anakinra and colchicine.
The case underscores the importance of considering somatic mosaicism in atypical or unresolved SAID cases.
Abstract
Neonatal-onset multisystem inflammatory disease (NOMID) and familial Mediterranean fever (FMF) are distinct entities within the expanding spectrum of systemic autoinflammatory diseases (SAIDs). We report a 3-month-old infant who presented with recurrent fever, urticarial rash, and polyarthritis. After excluding other causes, anakinra was initiated based on clinical suspicion of NOMID. Despite treatment optimisation, she continued to experience disease flares. An initial autoinflammatory panel and subsequent whole-exome sequencing revealed heterozygous MEFV (M694V and V726A) gene mutations, which did not explain the clinical picture. Further deep sequencing identified NLRP3 (p.Asp305Glu) somatic mosaicism, confirming NOMID. The coexistence of NOMID and FMF presented significant diagnostic and therapeutic challenges. Disease activity stabilised after colchicine was added. Clinicians…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
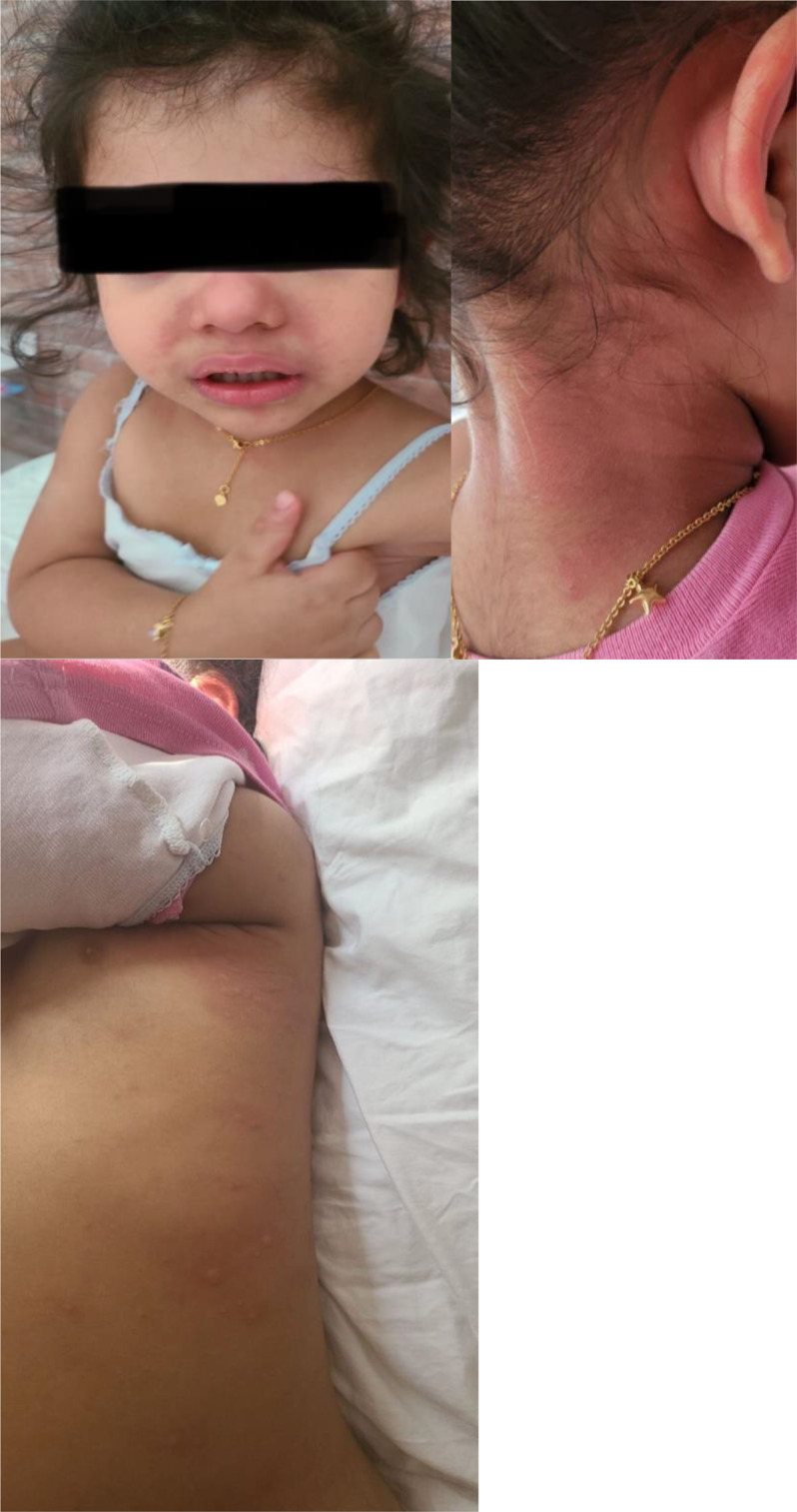 Fig. 1
Fig. 1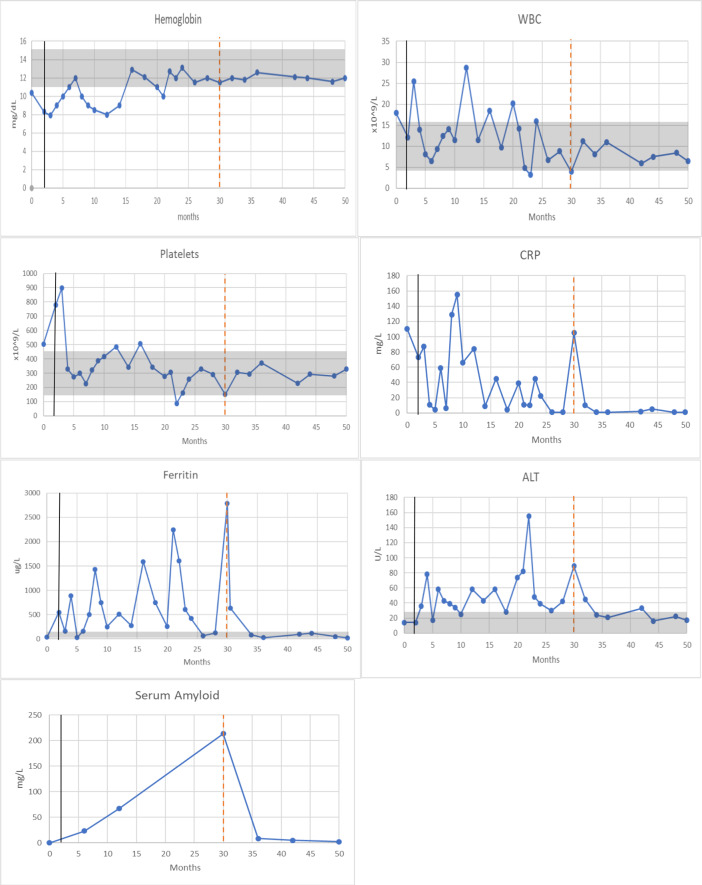 Fig. 2
Fig. 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInflammasome and immune disorders · Systemic Lupus Erythematosus Research · Urticaria and Related Conditions
1. Introduction
Cryopyrin-associated periodic syndrome (CAPS) is a dominantly inherited systemic autoinflammatory disease (SAID) comprising three overlapping clinical entities of varying severity: familial cold autoinflammatory syndrome (FCAS), Muckle–Wells syndrome (MWS) and neonatal-onset multisystem inflammatory disease (NOMID), with NOMID representing the most severe phenotype.^1^ Disease onset in NOMID may occur within hours of birth but is most commonly observed in neonates and infants. It is characterised by intermittent fever, urticarial rash, and persistently elevated acute-phase reactants. Neurological involvement, including chronic aseptic meningitis and papilledema, is typically evident at onset and may progress to brain atrophy, severe intellectual disability and hearing loss. Musculoskeletal manifestations such as hypertrophic arthropathy with contractures, frontal bossing and patellar overgrowth are also frequent.^2^
Detection of NLRP3 mutations is diagnostic in 65–70% of NOMID cases, includes both germline mutations and somatic mutations.^3^ While up to 40% of affected individuals test negative for germline NLRP3 mutations on conventional sequencing, but many are later found to carry somatic mosaicism.^3^ Multiple studies have identified a high incidence of somatic NLRP3 mosaicism in children diagnosed with NOMID.^3456^ Additionally, adult-onset disease has been reported in association with somatic NLRP3 mutations.^7^
Here, we report a case of NOMID due to somatic mosaicism in the NLRP3 gene presenting in early infancy. The diagnostic process was further complicated by the coexistence of another SAID, namely Familial Mediterranean Fever (FMF). To the best of the Authors' knowledge, this is the first reported case of two coexisting hereditary periodic fever syndromes associated with NLRP3 somatic mosaicism.
2. Case report
A previously healthy 3-month-old female infant presented with a 6-day history of high-grade fever reaching 40°C, irritability, generalised rash, bilateral conjunctivitis and swelling of the hands and feet. Clinical examination revealed a widespread, diffuse urticarial rash and warm wrists and ankles with restricted movement. Laboratory investigations showed leukocytosis, anaemia and elevated inflammatory markers (white blood cells [WBC] = 18 × 10^9^/L; haemoglobin [Hb] = 10 g/dL; platelets = 550 × 10^9^/L; C reactive protein [CRP] = 110 mg/L; erythrocyte sedimentation rate [ESR] = 60 mm/hr). Nasopharyngeal aspirates, viral screening, lumbar puncture and echocardiography (ECHO) were unremarkable. The child was initially managed as a case of incomplete Kawasaki disease with intravenous immunoglobulin (IVIG) and aspirin, in addition to cefazolin to cover potential bacterial infection. She showed clinical and biochemical improvement and was discharged.
Two weeks later, she re-presented with recurrence of fever, a diffuse urticarial rash, and widespread polyarthritis affecting the wrists, elbows, knees, ankles and hips. Differential diagnoses included systemic autoinflammatory diseases (SAIDs), systemic-onset juvenile idiopathic arthritis and haemophagocytic lymphohistiocytosis (HLH). A further history revealed a maternal nephew diagnosed with familial Mediterranean fever (FMF), carrying compound heterozygous mutations in the MEFV gene (M694V and V726A).
Laboratory evaluation again revealed leukocytosis, thrombocytosis and elevated inflammatory markers. Repeat ECHO showed a small rim of pericardial effusion and abdominal ultrasound revealed hepatosplenomegaly. HLH workup, including bone marrow examination, was normal. A skeletal survey showed no skeletal dysplasia, brainstem evoked response audiometry (BERA) was normal, and whole-body MRI revealed widespread polyarthritis with no intracranial abnormalities. Skin biopsy confirmed chronic urticarial changes. Empirical treatment was initiated with prednisolone (2 mg/kg) and anakinra (2 mg/kg) for a presumed SAID, specifically NOMID, pending genetic confirmation.
Three months later, an autoinflammatory gene panel identified a heterozygous pathogenic variant in the MEFV gene, c.2080A > G (p.Met694Val). Whole-exome sequencing (WES) subsequently identified an additional variant of uncertain significance (VUS) in the MEFV gene: p.Glu148Gln. The p.Glu148Gln (E148Q) variant in the MEFV gene is generally considered to have low penetrance and is often associated with a milder FMF phenotype when present alone. However, its pathogenicity may increase when combined with other mutations, such as p.Met694Val.^8^ Parental testing revealed that the father carried the p.Glu148Gln mutation and the mother carried the p.Met694Val variant in the MEFV gene; both were clinically asymptomatic.
Despite harbouring two heterozygous mutations in the MEFV gene, the patient's clinical picture was not consistent with FMF. Although prednisolone and anakinra were initiated for presumed NOMID, she continued to experience recurrent disease flares with high-grade fever, urticarial rash and arthritis, along with elevated inflammatory markers [Figures 1 and 2]. The anakinra dose was gradually increased to 6 mg/kg; however, the patient remained steroid-dependent and disease control remained suboptimal.
Images demonstrating disease flares manifested by fever associated with generalised urticarial rashes and irratibility.
Markers of inflammation in a neonatal-onset multisystem inflammatory diseases infant and her response to anakinra and colchicine. The x-axis is a time of disease course in months. Vertical black lines indicate the start of anakinra treatment, whereas dotted red lines represent the start of colchicine therapy. Gray shaded areas represent normal values. C reactive protein reference range is <1 mg/L. Serum Amyloid reference range is <10 mg/L. WBC = white blood cells; CRP = C-reactive protein; ALT = alanine transaminase.
As her symptoms did not align with typical FMF attacks despite WES results, including early onset, chronic urticarial rash and persistent arthritis, the search for alternative diagnoses continued. repeat WES with targeted deep sequencing was performed, revealing somatic mosaicism (5%) for a likely pathogenic variant in the NLRP3 gene (p.Asp305Glu), supporting a diagnosis of NOMID. The p.Asp305Glu variant is considered a novel mutation in NLRP3. Based on the combination of genetic and clinical findings, a diagnosis of concomitant NOMID and FMF was established. Colchicine was added to the treatment regimen to improve disease control and prevent further flares, resulting in a dramatic and sustained clinical improvement. Four years later, the child remains clinically stable, with normal growth and development and no evidence of central nervous system, skeletal, ocular or hearing impairment on routine screening.
3. Discussion
SAIDs are characterised by uncontrolled inflammation driven by the innate immune system. FMF is the first SAID to be described and remains the most common hereditary periodic fever syndrome. It is associated with mutations in the MEFV gene, which encodes the pyrin protein. The disease is most prevalent among populations living in the Mediterranean basin but has also been reported in other ethnic groups, including Japanese, Italians and Greeks.^89^
FMF typically presents during childhood, with attacks lasting one to three days. These episodes are characterized by fever, serositis, arthralgia and erysipelas-like skin lesions. In the current case, genetic testing identified two heterozygous variants in the MEFV gene. While the presence of compound heterozygous mutations may meet the genetic criteria for FMF, the clinical manifestations in our patient were atypical. The patient had prolonged febrile episodes exceeding 3 days, a persistent urticarial rash, hepatosplenomegaly and extensive polyarthritis beginning in early infancy—features more consistent with other monogenic autoinflammatory syndromes, such as NOMID.
NOMID is the rarest and most severe form of the CAPS.^1^ It is caused by mutations in the NLRP3 gene, which encodes cryopyrin. These mutations result in overactivation of the NLRP3 inflammasome and excessive production of interleukin-1 (IL-1), leading to a wide spectrum of inflammatory organ damage.^12^ Both classic and mosaic forms of NOMID share similar clinical features, though the severity and variability of symptoms may differ between the two.
Several reports have documented somatic NLRP3 mosaicism in patients with NOMID, highlighting that even a small proportion of mutated cells may suffice to initiate a sustained inflammatory response.^3456^ Conventional Sanger sequencing can detect mosaicism only when the mutation burden is relatively high.^10^
However, in CAPS—where mosaicism levels may be as low as 2% and over 100 disease-causing variants in NLRP3 have been reported—more sensitive techniques are often required. Deep sequencing allows for the detection of low-level mosaic mutations and may be necessary for accurate diagnosis in suspected cases of CAPS.
Tanaka et al. reported somatic NLRP3 mutations in 69.2% of patients with previously mutation-negative NOMID, suggesting that somatic mosaicism is a major contributor to disease pathogenesis.^3^ Their study found that patients with somatic NLRP3 mosaicism may present within the first 24 months of life and exhibit a disease phenotype as severe as that seen in germline mutation cases. Reported clinical features included fever (100%), urticarial rash (100%), meningitis (76%), arthritis (82%), walking disability (44%), intellectual disability (25%), and hearing loss (56%). In contrast, a UK centre described cases of NLRP3 mosaicism presenting with adult-onset disease, with a median age of 50 years (range: 31–71 years).^7^
Our patient, in whom 5% mosaicism for a likely pathogenic NLRP3 variant was detected, presented in early infancy but demonstrated a relatively mild disease course. This may be attributed to the early and aggressive initiation of IL-1 blockade and immunosuppressive therapy.
The IL-1 receptor is widely expressed across various tissues, making anakinra—an IL-1 receptor antagonist—an optimal therapeutic agent for inflammatory conditions. It blocks the binding of both IL-1α and IL-1β and has been employed in the treatment of a wide range of autoinflammatory and autoimmune disorders. Reports have highlighted the benefit of anakinra in patients who are refractory to standard therapies. Given the pivotal role of cryopyrin in regulating IL-1 release in patients with NLRP3 mutations, anti-IL-1 therapy remains the cornerstone of treatment in NOMID.^2^ Similarly, IL-1 blockade has shown efficacy in reducing the frequency of FMF attacks and mitigating subclinical inflammation.^11^
In this case, colchicine was not introduced initially due to the clinical focus on the diagnosis and management of NOMID. Since NOMID was considered the primary diagnosis and treatment for NOMID focuses on IL-1 inhibition, colchicine—the first-line therapy for FMF—was not prioritised. Although MEFV variants were identified, their significance was uncertain in the context of NOMID. While the maternal family history included a nephew with FMF, the patient lacked typical FMF features such as episodic fever or serositis at presentation, making a concurrent FMF diagnosis less evident at the time.
The patient's suboptimal response to anakinra alone prompted reconsideration of the diagnosis and raised the possibility of coexisting FMF. This led to the introduction of colchicine, resulting in improved disease control.
The combination of colchicine and anakinra was essential for managing coexisting disease activity and for reducing inflammatory markers, including serum amyloid levels. Anakinra functions by blocking IL-1 signaling, while colchicine suppresses inflammasome activation and inhibits microtubule polymerization, neutrophil chemotaxis, and adhesion. Together, these agents exert complementary anti-inflammatory effects, offering broader suppression of innate immune activation.^12^ The rationale for introducing colchicine despite the patient already receiving anakinra lies in its ability to target inflammasome activation upstream of IL-1 release. Following the initiation of colchicine, Fig. 2 demonstrates a clear and sustained reduction in both CRP and Serum Amyloid A levels, supporting its added anti-inflammatory effect alongside anakinra.
Moreover, colchicine has been shown to reduce the frequency and severity of FMF flares, some of which may not be fully suppressed by IL-1 blockade alone.^1314^ The therapeutic synergy between colchicine and anakinra in this patient led to a marked improvement in clinical stability. In retrospect, the delayed introduction of colchicine may also reflect the inherent diagnostic complexity of overlapping autoinflammatory syndromes, where clinical manifestations and inflammatory markers may be attributed to either disease, complicating initial therapeutic decisions and prioritisation.
The coexistence of two autoinflammatory diseases in one patient is rare but has been reported.^151617^
Although uncommon, the occurrence of more than one autoinflammatory disease in a single patient is a recognised clinical phenomenon. Most previously documented cases have involved tumour necrosis factor receptor-associated periodic syndrome and FMF; however, this appears to be the first reported case involving FMF and NOMID associated with NLRP3 somatic mosaicism.
4. Conclusion
This case presented both diagnostic and therapeutic challenges, emphasising the need for a high index of clinical suspicion, as systemic autoinflammatory diseases often exhibit overlapping clinical features. Clinicians must remain alert to atypical symptoms that may suggest the presence of concurrent syndromes. The limitations of conventional DNA sequencing highlight the importance of employing next-generation sequencing techniques to detect mosaic variants in suspected cases. Ultimately, early recognition and prompt initiation of appropriate treatment are essential for improving outcomes and prognosis in patients with complex autoinflammatory conditions.
Authors' Contribution
Eman Al Masroori: Data Curation, Writing - Original Draft, Review, Validation. Mahadev Mal: Data Curation, Review & Editing, Validation. Reem Abdwani: Conceptualization, Methodology, Validation, Supervision, Writing - Review & Editing.
Ethics Statement
Consent was obtained from the patient's parent/guardian for the publication of this case report.
Data Availability Statement
Data is available upon reasonable request from the corresponding author.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Goldbach-Mansky R. Current status of understanding the pathogenesis and management of patients with NOMID/CINCA. Curr Rheumatol Rep 2011; 13:123–31. https://doi.org/10.1007/s 11926-011-0165-y.10.1007/s 11926-011-0165-y 21538043 PMC 3195512 · doi ↗ · pubmed ↗
- 2Finetti M Omenetti A Federici S Caorsi R Gattorno M. Chronic Infantile Neurological Cutaneous and Articular (CINCA) syndrome: A review. Orphanet J Rare Dis 2016; 11:167. https://doi.org/10.1186/s 13023-016-0542-8.10.1186/s 13023-016-0542-827927236 PMC 5142346 · doi ↗ · pubmed ↗
- 3Tanaka N Izawa K Saito MK Sakuma M Oshima K. High incidence of NLRP 3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: Results of an international multicenter collaborative study. Arthritis Rheum 2011; 63:3625–32. https://doi.org/10.1002/art.30512.10.1002/art.3051221702021 PMC 3498501 · doi ↗ · pubmed ↗
- 4Saito M Nishikomori R Kambe N Fujisawa A Tanizaki H Takeichi K. Disease-associated CIAS 1 mutations induce monocyte death, revealing low-level mosaicism in mutation-negative cryopyrin-associated periodic syndrome patients. Blood 2008; 111:2132–41. https://doi.org/10.1182/blood-2007-06-094201.10.1182/blood-2007-06-09420118063752 · doi ↗ · pubmed ↗
- 5Omoyinmi E Melo Gomes S Standing A Rowczenio DM Eleftheriou D Klein N. Whole-exome sequencing revealing somatic NLRP 3 mosaicism in a patient with chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheumatol 2014; 66:197–202. https://doi.org/10.1002/art.38217.10.1002/art.3821724431285 PMC 3995009 · doi ↗ · pubmed ↗
- 6Aróstegui JI Lopez Saldaña MD Pascal M Clemente D Aymerich M Balaguer F. A somatic NLRP 3 mutation as a cause of a sporadic case of chronic infantile neurologic, cutaneous, articular syndrome/neonatal-onset multisystem inflammatory disease: Novel evidence of the role of low-level mosaicism as the pathophysiologic mechanism underlying Mendelian inherited diseases. Arthritis Rheum 2010; 62:1158–66. https://doi.org/10.1002/art.27342.10.1002/art.2734220131270 · doi ↗ · pubmed ↗
- 7Rowczenio DM Gomes SM Aróstegui JI Mensa-Vilaro A Omoyinmi E Trojer H. Late-onset cryopyrin-associated periodic syndromes caused by somatic NLRP 3 mosaicism: UK single-center experience. Front Immunol 2017; 8:1410. https://doi.org/10.3389/fimmu.2017.01410.10.3389/fimmu.2017.0141029163488 PMC 5671490 · doi ↗ · pubmed ↗
- 8Orouk Awaad E Khoury Lvan Straalen JW Miller-Barmak A Gazitt T Haddad-Haloun J Brik R Hamad Saied M. E 148Q variant: A familial mediterranean fever-causing mutation or a sequence variant? Eur J Pediatr 2024 Oct; 183(10):4499–4506. doi: 10.1007/s 00431-024-05690-5.39143349 PMC 11413036 · doi ↗ · pubmed ↗