Clinicopathological Spectrum of Myxoid Soft Tissue Tumours with Emphasis on Diagnostic Differentials
Sanjana Ahuja, Nisha Modi, Amit Varma, Shivani Kapur, Kamal Malukani, Harshita Deep Sahu

TL;DR
This study examines the characteristics and diagnostic challenges of myxoid soft tissue tumors, focusing on their clinical and morphological features to improve accurate diagnosis.
Contribution
The study provides a detailed clinicopathological analysis of myxoid soft tissue tumors from a specific geographic region, emphasizing diagnostic differentials.
Findings
Benign myxoid soft tissue tumors were more common (60.9%) and predominantly occurred in the upper limb and younger age group.
Myxoid schwannoma was the most prevalent benign tumor, while myxofibrosarcoma was the most frequent malignant tumor.
A statistically significant correlation was found between tumor grade and size or depth in malignant cases.
Abstract
Myxoid soft tissue tumours (MSTTs) represent a complex group of mesenchymal neoplasms characterised by the production of an extracellular myxoid matrix, which often poses diagnostic challenges. This study aimed to identify the relative frequency, distribution and morphological spectrum of MSTT as well as to illustrate commonly encountered diagnostic difficulties. This retrospective study of the clinicopathological profiles of MSTT cases at Sri Aurobindo Medical College & Post Graduate Institute, Indore, India, was conducted from January 2020 to December 2023. A total of 110 MSTT cases were included in this study. These cases accounted for 7.3% of the soft tissue tumours (STTs), with a predominance of benign MSTT (n = 67, 60.9%). The fibroblastic/myofibroblastic family of STT constituted the majority (33.6%). The upper limb (34%) was the most frequently involved anatomical site for…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
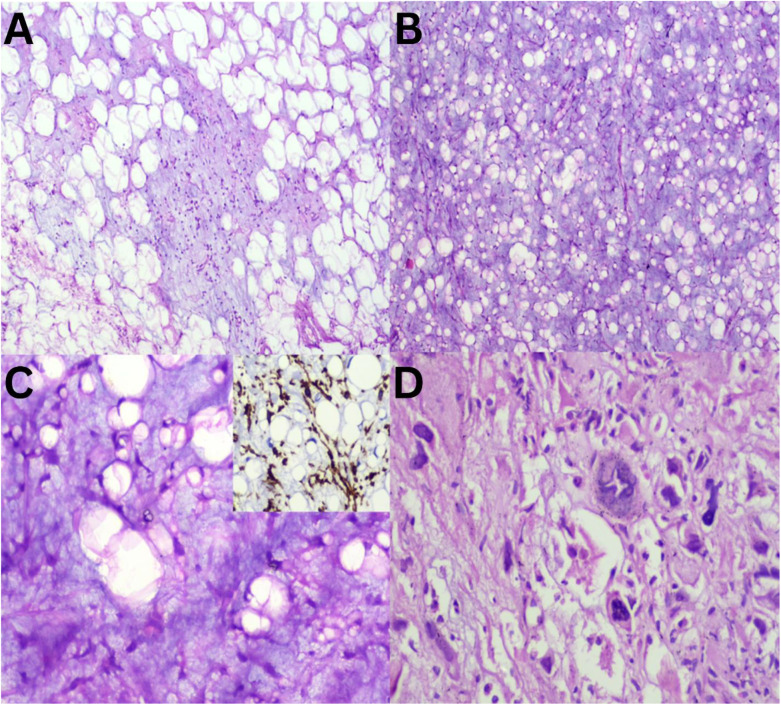 Fig. 1
Fig. 1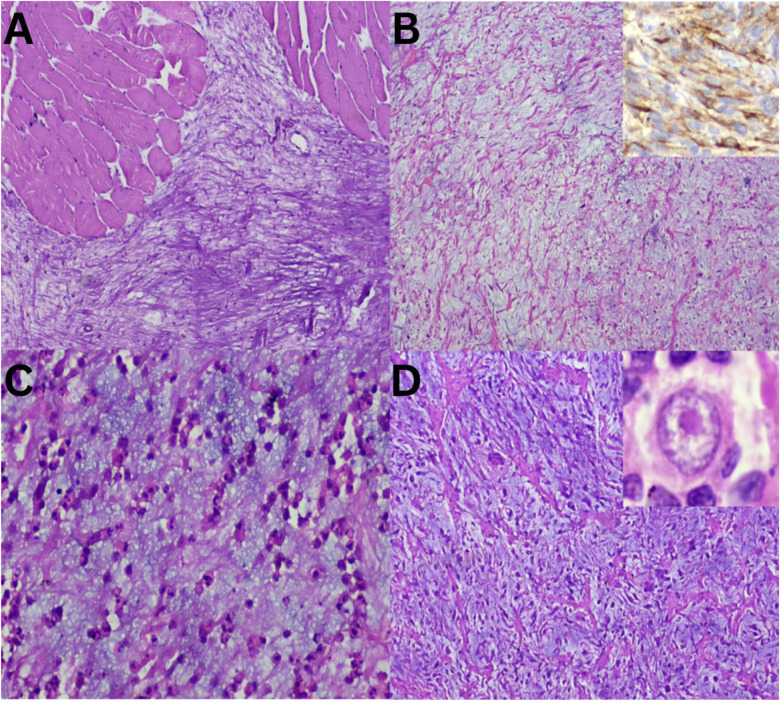 Fig. 2
Fig. 2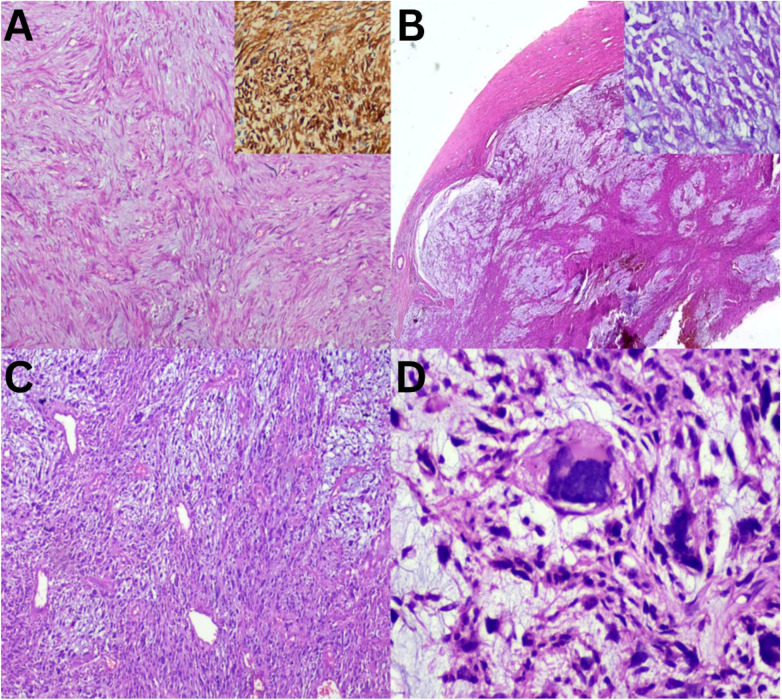 Fig. 3
Fig. 3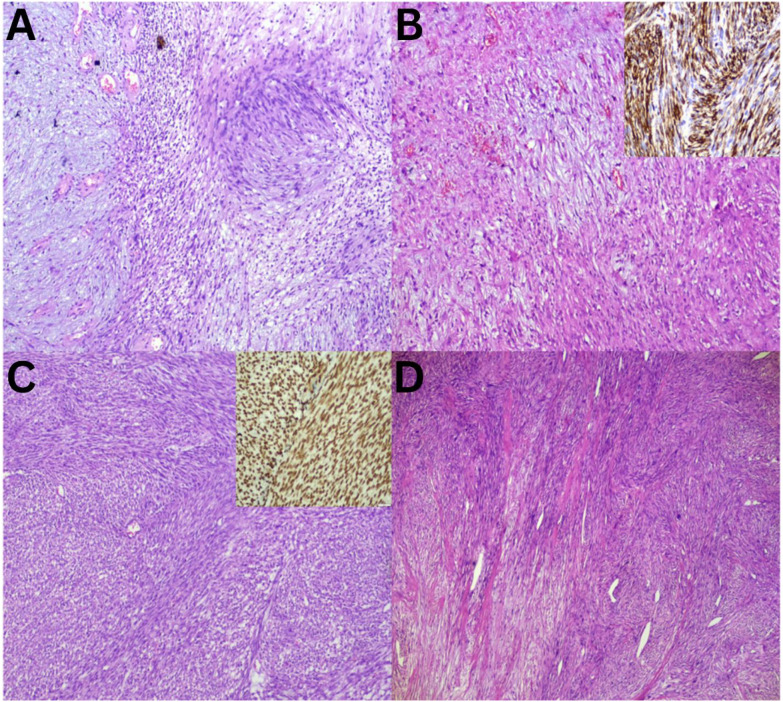 Fig. 4
Fig. 4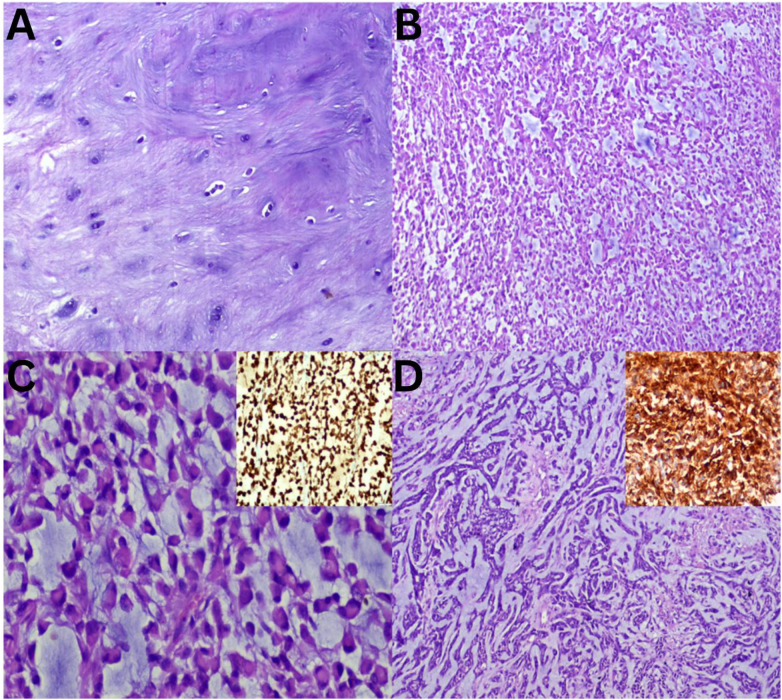 Fig. 5
Fig. 5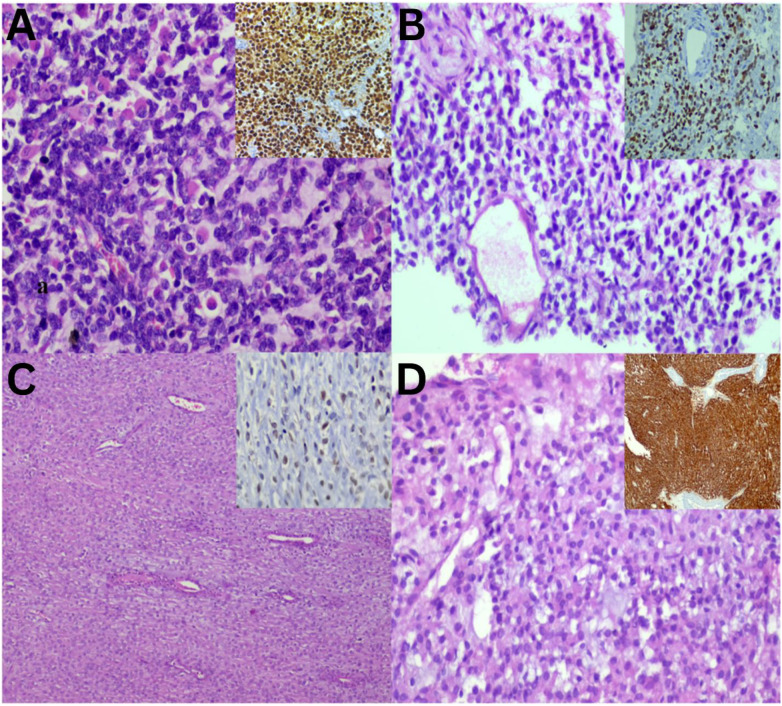 Fig. 6
Fig. 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMusculoskeletal synovial abnormalities and treatments · Sarcoma Diagnosis and Treatment · Nail Diseases and Treatments
1. Introduction
Myxoid soft tissue tumours (MSTTs) represent a diverse category of mesenchymal tumours characterised by the presence of extracellular myxoid material.^1^ Glycosaminoglycans or mucopolysaccharides are the predominant constituents of this myxoid substance.^2^ MSTTs comprise a spectrum of lesions with diagonally opposite clinical implications, ranging from benign to malignant neoplasms.^3^ Additionally, some common epithelial tumours may show myxoid changes upon histological examination. Diagnostic challenges arise in the categorisation of myxoid neoplasms due to their deceptive appearance, which can easily lead to misidentification with similar conditions. This difficulty is attributed to appreciable clinical, radiologic, cytologic and histomorphological overlap as well as intratumoural variability. Additionally, the limited utility of immunohistochemistry (IHC) and molecular studies further complicates accurate assessment.
This study aimed to highlight the most common types of MSTT, describe their clinicopathological characteristics and explore the differential diagnoses that may confound even the most seasoned pathologists. It emphasises the pivotal role of keen morphological observation and precise diagnostic acumen.
2. Methods
This retrospective study was conducted from January 2020 to December 2023 at the Department of Pathology, Sri Aurobindo Medical College & Post Graduate Institute, Indore, India. A convenience sampling method was used due to the relative rarity of MSTT cases.
Pathology records of all diagnosed soft tissue tumours (STTs), including those confirmed by IHC, were retrieved. Haematoxylin and eosin stained slides and IHC slides were thoroughly reviewed and reclassified as MSTTs based on the presence of the myxoid component. Cases were further sub-grouped as benign (including the intermediate category) or malignant, according to different families outlined in World Health Organization classificationof STTs.^1^ This includes adipocytic, fibroblastic/ myofibroblastic, vascular, smooth muscle, skeletal muscle, gastrointestinal stromal tumours (GISTs), chondro-osseous tumours, peripheral nerve sheath tumours, tumours of uncertain differentiation and undifferentiated small round cell tumours (SRCTs).
STTs may show a variable amount of myxoid components, and there are different types of myxoid lesions. Some tumours, such as myxofibrosarcoma (MFS), have a myxoid presence as an integral part of their diagnosis. Conversely, other tumours, which are not usually associated with myxoid changes, may still demonstrate a considerable amount of myxoid material; for example, GIST can exhibit myxoid changes. This study encompassed both categories of tumours. However, STTs classified as MSTT were only those that, upon morphological analysis, revealed a 20% myxoid component or greater.
Patients eligible for inclusion in this study were those with STT who met the criteria for MSTT defined above. The included cases consisted of resection specimens with tumours that provided sufficient histological material for detailed analysis. Additionally, the inclusion criteria mandated that cases had comprehensive and adequate clinical details available for thorough examination.
On the other hand, cases of STT were excluded from the study if they contained a myxoid component of less than 20%. Furthermore, cases lacking sufficient histological material for proper analysis and those without essential clinical details or epithelial tumours exhibiting myxoid differentiation were also excluded. Additionally, cases reported from core biopsies or those that had relapsed were not considered for inclusion in this study.
Data were analysed using the Statistical Package for the Social Sciences (SPSS) software, Version 16 (IBM Corp. Armonk, New York, USA). Category variables were depicted as frequencies and percentages. Intergroup comparisons were conducted using the Chi-square test, with a P value of <0.05 considered statistically significant.
3. Results
A total of 1,552 STTs were reported during the study period. Of these, 45 cases were excluded due to inadequate clinical information/histological material. Consequently, 1,507 cases were reviewed and reclassified based on the MSTT defined criteria which accounted for 110 cases (7.3%) of the total, with a male predominance of 61.8% (n = 68) among both the benign (including intermediate) and malignant groups.
These MSTTs were segregated into sub-families according to the WHO classification: adipocytic (n = 15, 13.6%), fibroblastic/myofibroblastic (n = 37, 33.6%), smooth muscle (n = 11, 10%), skeletal muscle (n = 1, 0.9%), GIST (n = 4, 3.6%), chondro-osseous (n = 1, 0.9%), peripheral nerve sheath (n = 14, 12.7%), tumours of uncertain differentiation (n = 25, 22.7%) and undifferentiated SRCTs (n = 2, 1.8%).^1^
The fibroblastic/myofibroblastic family of STT represented the largest proportion (n = 37, 33.6%). Myxoid schwannoma (n = 9, 13.4%) was the most prevalent benign tumour, followed by myxoid fibromatosis (n = 8, 11.9%) and myxoid leiomyoma (n = 8, 11.9%). Other entities within various families included myxolipoma (n = 5), myxoid nodular fasciitis (n = 4), myxoid dermatofibrosarcoma protuberans (n = 4), solitary fibrous tumour with myxoid change (n = 3), myxoid inflammatory myofibroblastic tumour (IMFT; n = 2), myxoid GIST (n = 4), soft tissue chondroma (n = 1), myxoid neurofibroma (n = 2), dermal nerve sheath myxoma (n = 1), intramuscular myxoma (n = 6), juxta-articular myxoma (n = 1), ganglion cyst (n = 6) and myoepithelioma with myxoid change (n = 3).
MFS (n = 9, 20.9%) was the most frequently observed malignancy, followed by liposarcoma (n = 8, 18.6%) myxoinflammatory fibrosarcoma (MIFS; n = 4, 9.3% and undifferentiated sarcoma (n = 4, 9.3%In addition, several other malignant tumours from various families were identified, including infantile fibrosarcoma with myxoid change (n = 1), adult fibrosarcoma with myxoid change (n = 2), low-grade fibromyxoid sarcoma (LGFS; n = 1), leiomyosarcoma (LMS) with myxoid change (n = 3), rhabdomyosarcoma (RMS) with myxoid change (n = 1), malignant peripheral nerve sheath tumour with myxoid change (n = 2), synovial sarcoma with myxoid change (n = 1), clear cell sarcoma (CCS) with myxoid change (n = 1), myoepithelial carcinoma (MEC) with myxoid change (n = 2), extraskeletal myxoid chondrosarcoma (EMC; n = 3) and extraskeletal Ewing sarcoma (EES; n = 2).
The upper limb (n = 23, 34%) was the most frequently affected anatomical site in benign MSTT, with the 20–40 age range being the most common (46.3%). Tumour sizes were predominantly below 5 cm (57%), with 84% being superficial locations. In malignant MSTT, the lower limb (n = 21, 49%) was largely affected, with the 40–60 age group being the most prevalent (46.5%). The tumour size range of 5–10 cm was most common (46.5%), with the majority located deep (84%).
All malignant sarcomas included in this study were graded based on the National Federation of Comprehensive Cancer Centres system (i.e. Fédération Nationale des Centres de Lutte Contre le Cancer).^1^ Of all 43 cases examined, 7 (16.3%) were grade I, 12 (27.9%) were grade II and the remaining were categorised as grade III. A statistically significant correlation was observed between the histological grade of the tumour and its size (P = 0.006) as well as between the grade and the depth of the tumour (P = 0.015). Overall, 56% of the malignancies were grade III tumours, which were predominantly larger than 10 cm (n = 14, 70%) and located deep (n = 23, 64%).
4. Discussion
Myxoid lesions represent a group of mesenchymal entities characterised by divergent differentiation and the presence of a myxoid matrix. Although many STTs can exhibit myxoid changes, this feature is not prominent in all cases and may be regarded as a ‘non-myxoid’ tumour that can demonstrate myxoid change. In this study, the incidence of MSTTs was 7.3%, which is higher than the 3.7% reported by Sigamani and Ponnuswamy.^4^ The probable reason for this discrepancy could be the inclusion of ‘non-myxoid’ STT, where myxoid change is not usually seen but may develop myxoid differentiation or have myxoid variants, such as LMS with myxoid change.
Benign MSTTs (60.9%) outnumbered malignant sarcomas (39%), which were comparable to the overall distribution of STT as reported by the WHO.^1^ However, it contrasts with Sigamani and Ponnuswamy findings, which indicated that sarcomas surpassed benign MSTT.^4^ Additionally, the current study observed a male gender preponderance, consistent with previously reported data.^4^
When comparing the distribution of benign and malignant MSTT with other studies, this study found that the fibroblastic/myofibroblastic family of STT represented the largest proportion (31.3%) among benign MSTT, which aligns with the findings of Kransdorf.^5^ However, this differs from the studies conducted by Yuceturk et al. and Sigamani and Ponnuswamy who reported vascular (37.5%) and peripheral nerve sheath tumours (47.8%) as the predominant categories, respectively.^46^ Similarly, regarding malignant sarcomas, this study observed that the fibroblastic/myofibroblastic group accounted for the maximum number of cases (37.2%) resonating with the reports by Sigamani and Ponnuswamy (40.6%).^4^ Conversely, tumours of uncertain differentiation were predominant in the studies by Yuceturk et al. (37.5%) and Coindr et al. (38.2%).^67^
Myxoid lesions exhibit a complex spectrum showcasing varying cellularity coupled with a variable amount of myxoid substance. Consequently, differentiating these tumours poses a diagnostic puzzle in several instances.^89^ Additionally, their clinical behaviour and prognosis differ significantly.^10^ There overlapping morphological features of adipocytic tumours, where myxolipoma and myxoid liposarcoma (MLPS) both contain abundant mature fat [Figure 1A and B]. However, upon closer examination, MLPS reveals signet ring cell lipoblasts and crow's feet vasculature, whereas pleomorphic liposarcoma (PLPS) evidently displays marked nuclear atypia [Figure 1C and D]. These lesions have contrasting biological natures. Some myxoid lesions, such as myxoma, show abundant myxoid material. Hypocellular myxoid lesions, including intramuscular myxoma resembles an IMFT at low magnification [Figure 2A and B]. However, at a higher power, the presence of rich inflammatory infiltrates and myofibroblastic proliferation becomes evident [Figure 2C]. In the aforementioned category of myxoid lesions, myxoinflammatory fibroblastic sarcoma (MIFS) can be incorporated [Figure 2D]. This entity is characterised by the presence of interspersed large cells with macronucleoli within a relatively cellular myxoid stroma. Within the realm of lesions exhibiting abundant extracellular myxoid matrix, the current study identified cases ranging from benign myxoma to malignant MIFS, with IMFT exhibiting borderline behaviour.
Adipocytic myxoid soft tissue tumours sections. A: Haematoxylin and eosin (H & E) staining at ×20 magnification showing myxolipoma with abundant mature fat and myxoid areas. B & C: H & E staining at ×20 and ×40 magnifications, respectively, showing myxoid liposarcoma with signet ring type lipoblast and (inset = diaminobenzidine at ×40 magnification) capillary vasculature highlighted by CD34. D: H & E staining at ×40 magnification showing pleomorphic liposarcoma with bizarre forms.
Soft tissue tumours with abundant myxoid matrix sections. A: Haematoxylin and eosin (H & E) staining at ×20 magnification showing intramuscular myxoma. B: H & E staining at ×20 magnification showing inflammatory myofibroblastic tumour with spindle myofibroblast highlighted by (inset = diaminobenzidine at ×40 magnification) positive ALK immunostain. C: H & E staining at ×40 magnification showing numerous inflammatory cells. D: H & E staining at ×40 magnification showing myxoinflammatory fibroblastic sarcoma with large cells and myxoid stroma with (inset) macronucleoli.
Moving from hypocellular to relatively cellular myxoid lesions, LGFS, characterised by bland spindle cells arranged in small fascicles and MFS, exhibits alternating hypocellular myxoid and hypercellular areas [Figure 3A and B]. MFS is notable for its prominent curvilinear vasculature and the presence of pleomorphic giant cells [Figure 3C and D]. In the current study, MFS was the most frequently observed MSTT, consistent with findings reported in previous literature.^4^ STT, referred to as a ‘non-myxoid’ tumour, is characterised by the absence of a prominent myxoid matrix, although myxoid changes may develop [Fig. 4]. Schwannomas may exhibit myxoid changes; however, the presence of obvious Verocay bodies facilitates diagnosis [Figure 4A]. Conversely, other similar-looking spindle cell tumours within this category, such as LMS, synovial sarcoma and fibrosarcoma, cannot be differentiated based solely on morphology [Figure 4B–D]. Therefore, it is essential to subject these tumours to a series of lineage-specific immunomarkers to achieve a definitive diagnosis, which will aid in the accurate selection of the appropriate management modality.^1112^
Relatively cellular myxoid soft tissue tumour sections. A: Haematoxylin and eosin (H & E) staining at ×20 magnification showing low-grade fibromyxoid sarcoma with bland spindle cells in short fascicles stained positive for MUC4 (inset = diaminobenzidine at ×20 magnification). B: H & E staining at ×10 magnification showing low-grade myxofibrosarcoma showing multinodularity with (inset = H & E staining at ×40 magnification) hyperchromatic fibroblast nuclei. C: H & E staining at ×20 magnification showing high-grade myxofibrosarcoma with prominent, curvilinear vasculature. D: H & E staining at ×40 magnification showing pleomorphic giant cells.
Sections of so-called ‘non-myxoid’ soft tissue tumours with myxoid component and cellular fascicular arrangement. A: Haematoxylin and eosin (H & E) staining at ×20 magnification showing schwannoma with Verocay bodies. B: H & E staining at ×20 magnification showing leiomyosarcoma with positive immunostaining for H-caldesmon (inset = diaminobenzidine at ×20 magnification). C: H & E staining at ×20 magnification showing synovial sarcoma with positive immunostaining for TLE1 (inset = diaminobenzidine at ×20 magnification). D: H & E staining at ×20 magnification showing fibrosarcoma with herringbone pattern.
In addition to these findings, the current study identified several cases exhibiting chondromyxoid stroma [Fig. 5]. At the benign end of this spectrum, soft tissue chondroma is characterised by the presence of chondrocytes within a chondromyxoid matrix [Figure 5A]. Conversely, the malignant end is illustrated by EMC, which is cellular and shows rhabdoid-like cells [Figure 5B and C]. MEC demonstrates that both EMC and MEC share a similar low-power view [Figure 5D]. Mandatory IHC confirms their respective diagnoses. STT can present with round cell morphology and myxoid changes. Each of these entities warrants divergent treatment approaches and prognostic considerations. RMS can be distinguished from the others due to the striking appearance of rhabdomyoblasts [Figure 6A]. A case of EES shows uniform round cells with myxoid stroma, resembling a case of CCS of soft tissue and GIST [Figure 6B–D]. The primary noticeable difference among these tumours are that EES tumour cells exhibit clear cytoplasm and a perivascular arrangement, whereas CCS shows epithelioid cells arranged in compact nests, and GIST displays epithelioid cells with eosinophilic cytoplasm. Positive immunostaining for NKX2.2 [Figure 6B inset] in EES, SOX10 [Figure 6C inset] in CCS and DOG1 [Figure 6D inset] in GIST substantiates the specific diagnoses and underscores the importance of IHC in selected cases.
Soft tissue tumours with chondromyxoid matrix sections. A: Haematoxylin and eosin (H & E) staining at ×40 magnification showing soft tissue chondroma showing chondrocytes surrounded by a hyaline matrix. B: H & E staining at ×20 magnification showing extraskeletal myxoid chondrosarcoma with tumour cells in cords. C: H & E staining at ×40 magnification showing rhabdoid-looking cells in extraskeletal myxoid chondrosarcoma with positive NSE (inset = diaminobenzidine at ×20 magnification). D: H & E staining at ×20 magnification showing myoepithelial carcinoma with myxoid areas and areas of necrosis and positive vimentin (inset = diaminobenzidine at ×20 magnification).
Soft tissue tumours with round cell morphology and myxoid component sections. A: Haematoxylin and eosin (H & E) staining at ×40 magnification showing rhabdomyosarcoma with rhabdomyoblast positive for Myogenin (inset = diaminobenzidine at ×20 magnification). B: H & E staining at ×40 magnification showing extraskeletal Ewing sarcoma with the perivascular arrangement and immunopositivity for NKX2.2 (inset = diaminobenzidine at ×20 magnification). C: H & E staining at ×20 magnification showing clear cell sarcoma with epithelioid cells and positive SOX10 (inset = diaminobenzidine at ×40 magnification). D: H & E staining at ×40 magnification showing epithelioid GIST with positive DOG1 (inset = diaminobenzidine at ×20 magnification).
Importantly, while MFS is the most common variant of MSTT, there is no confirmatory ancillary technique for diagnosis, which is made by exclusion. Nonetheless, differential diagnoses for high-grade MFS may include a set of high-grade spindle cell sarcomas with myxoid differentiation like PLPS, LMS and EMC.^131415^ Low-grade MFS may be confused with LGFS, fibromatosis, nodular fasciitis and schwannoma with a myxoid matrix.^1617^ An exhaustive literature search also accentuates instances where myxoid GIST has been mislabelled as myxoid LMS, consequently leading to erroneous treatment in conditions that require targeted therapy.^18^ Furthermore, multiple exemplifications of diagnostic pitfalls have been documented, underscoring the urgent need to recognise the heterogeneity present in myxoid neoplasms.^192021^
The role of IHC in the evaluation of STT remains crucial. Traditionally, the primary purpose of IHC in this field was to establish a line of differentiation; however, many conventional markers, such as smooth muscle actin and S100 protein, have demonstrated limited specificity.^22^ Fortunately, over the past decade, more specific immunohistochemical markers for STT have been developed, thereby enhancing diagnostic accuracy.^23^ Molecular studies play a pivotal role in improving the understanding, diagnosis and management of STT. As technology continues to evolve and the knowledge of genetic alterations expands, the incorporation of molecular testing into clinical practice will increasingly be vital for delivering personalised care to patients with STT.^2425^
5. Conclusion
This study provides a comprehensive array of clinical and morphological profiles of MSTT, each with discrete clinical implications. Additionally, it highlights the significance of understanding several MSTT differentials, which are influenced by variations in cellularity and the amount of myxoid stroma. Furthermore, this study emphasises the vital role of meticulous morphological observation and reinforces the value of deciphering immunostaining results within the context of histology.
Funding
No funding was received for this study.
Ethics Statement
Ethical approval was obtained from the institutional review board and ethics committee of Sri Aurobindo Medical College & Post Graduate Institute for this observational, retrospective and single-centre study. A waiver of consent was granted for this research.
Conflict of Interest
The authors declare that there are no conflicts of interest.
Data Availablility
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Authors' Contributions
SA contributed to manuscript writing, manuscript editing and data acquisition. NM contributed to manuscript writing, photography, manuscript editing, immunohistochemistry interpretation and histopathology reporting. AV conducted a critical review of the manuscript's scientific content of as well as manuscript editing and histopathology reporting. SK and HDS contributed to the compilation of photographs, literature review and manuscript writing. KM provided expert opinions and conducted a critical review of the scientific content of the manuscript. All authors approved the final version of the manuscript.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World Health Organization. The WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours, 5th ed. Lyon, France:International Agency for Research on Cancer (IARC), 2020.
- 2Willems SM Wiweger Mvan Roggen JF Hogendoorn PC. Running GA Gs: Myxoid matrix in tumor pathology revisited: What's in it for the pathologist? Virchows Arch 2010; 456:181–92. https://doi.org/10.1007/s 00428-009-0822-y.10.1007/s 00428-009-0822-y 19705152 PMC 2828560 · doi ↗ · pubmed ↗
- 3Graadt van Roggen JF Hogendoorn PC Fletcher CD. Myxoid tumours of soft tissue. Histopathology 1999; 35:291–312. https://doi.org/10.1046/j.1365-2559.1999.00835.x.10.1046/j.1365-2559.1999.00835.x 10564384 · doi ↗ · pubmed ↗
- 4Sigamani K Ponnuswamy K. Clinicopathological profile of myxoid soft tissue tumors- A retrospective study in a tertiary care hospital in South India. Niger J Clin Pract 2022; 25:1584–92. https://doi.org/10.4103/njcp.njcp_292_22.10.4103/njcp.njcp_292_2236149223 · doi ↗ · pubmed ↗
- 5Kransdorf MJ. Benign soft-tissue tumors in a large referral population: Distribution of specific diagnoses by age, sex, and location. AJR Am J Roentgenol 1995; 164:395–402. https://doi.org/10.2214/ajr.164.2.7839977.10.2214/ajr.164.2.78399777839977 · doi ↗ · pubmed ↗
- 6Yücetürk G Sabah D Keçeci B Kara AD Yalçinkaya S. Prevalence of bone and soft tissue tumors. Acta Orthop Traumatol Turc 2011; 45:135–43. https://doi.org/10.3944/AOTT.2011.2504.10.3944/AOTT.2011.250421765225 · doi ↗ · pubmed ↗
- 7Coindre JM Terrier P Guillou L Le Doussal V Collin F Ranchère D. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: A study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001; 91:1914–26. https://doi.org/10.1002/1097-0142(20010515)91:103.0.co;2-3.10.1002/1097-0142(20010515)91:103.0.co;2-311346874 · doi ↗ · pubmed ↗
- 8Layfield LJ Dodd L Klijanienko J. Myxoid neoplasms of bone and soft tissue: A pattern-based approach. J Am Soc Cytopathol 2021; 10:278–92. https://doi.org/10.1016/j.jasc.2020.09.009.10.1016/j.jasc.2020.09.00933168472 · doi ↗ · pubmed ↗