The genome sequence of the Slender-footed Robberfly, Leptarthrus brevirostris (Meigen, 1804)
Robert Wolton, Tianzhu Xiong, Ken Kraaijeveld

TL;DR
This paper presents the genome sequence of the Slender-footed Robberfly, including detailed assemblies of its haplotypes and mitochondrial genome.
Contribution
The study provides a high-quality genome assembly for Leptarthrus brevirostris, including chromosomal pseudomolecules and mitochondrial DNA.
Findings
The genome assembly includes two haplotypes totaling over 850 megabases each.
Haplotype 1 is mostly scaffolded into six chromosomal pseudomolecules.
The mitochondrial genome is 18.16 kilobases in length.
Abstract
We present a genome assembly from a female specimen of Leptarthrus brevirostris (Slender-footed Robberfly; Arthropoda; Insecta; Diptera; Asilidae). The assembly contains two haplotypes with total lengths of 850.81 megabases and 851.46 megabases. Most of haplotype 1 (99.05%) is scaffolded into 6 chromosomal pseudomolecules. Haplotype 2 was assembled to scaffold level. The mitochondrial genome has also been assembled, with a length of 18.16 kilobases.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1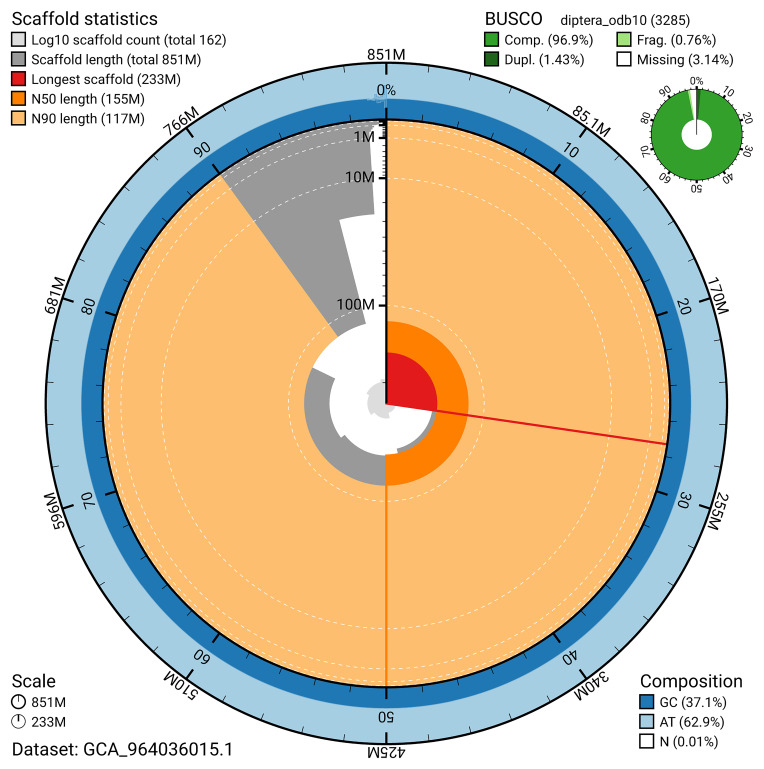 Figure 2
Figure 2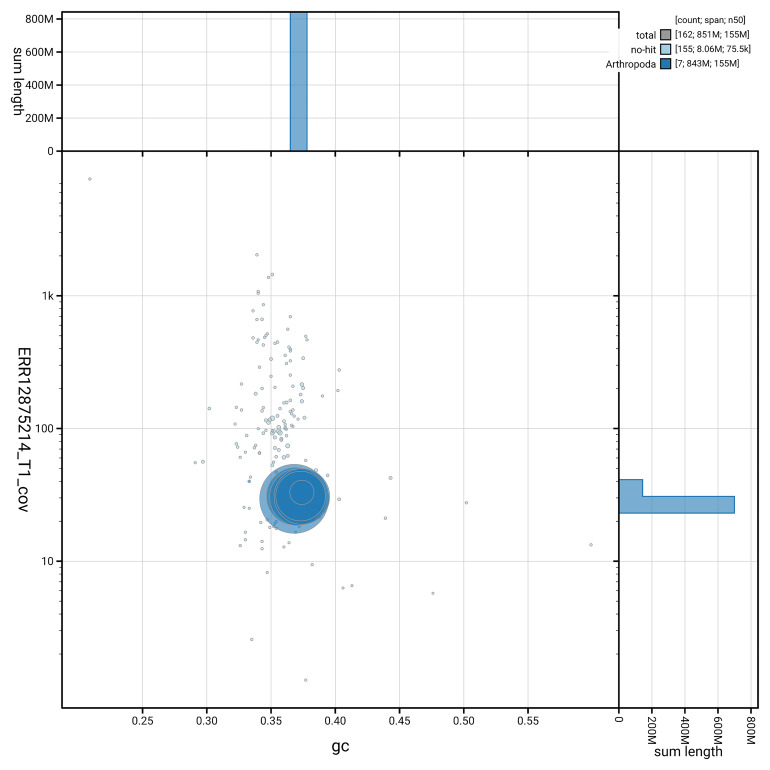 Figure 3
Figure 3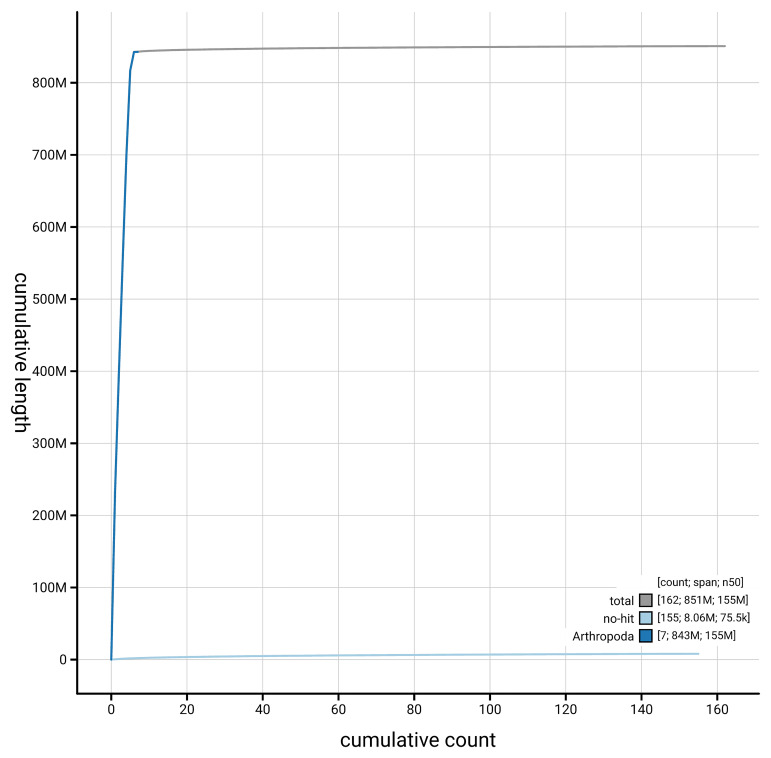 Figure 4
Figure 4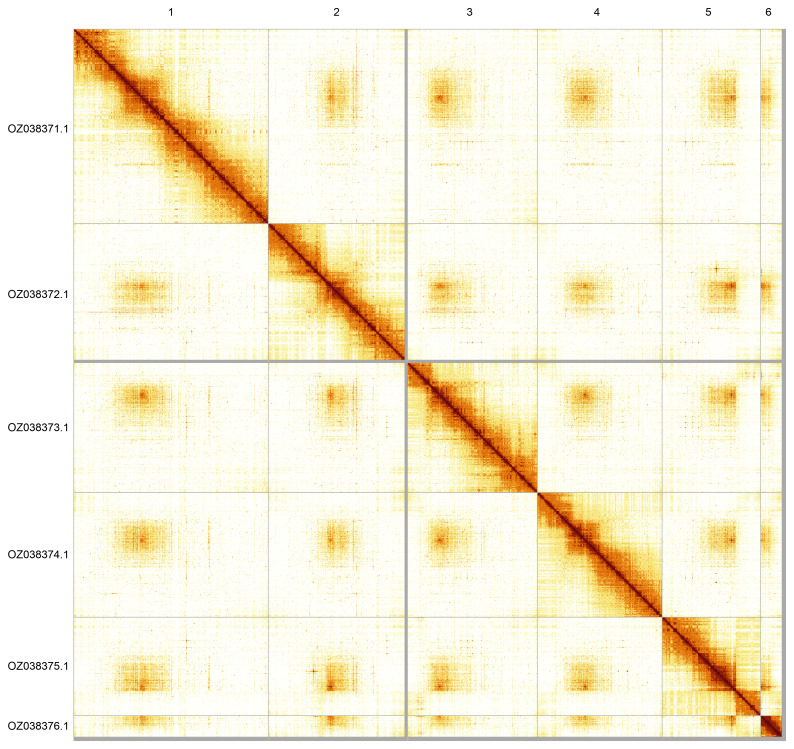 Figure 5
Figure 5| Project information | |||
|---|---|---|---|
|
| Leptarthrus brevirostris (slender-footed robberfly) | ||
|
| PRJEB74689 | ||
|
|
| ||
|
| SAMEA112222057 | ||
|
| 468750 | ||
| Specimen information | |||
|
|
|
|
|
|
| idLepBrev1 | SAMEA112222157 | thorax and abdomen |
|
| idLepBrev1 | SAMEA112222379 | head |
|
| idLepBrev1 | SAMEA112222157 | thorax and abdomen |
| Sequencing information | |||
|
|
|
|
|
|
| ERR12893022 | 1.02e+09 | 154.29 |
|
| ERR12875214 | 3.04e+06 | 25.8 |
|
| ERR12893023 | 6.79e+07 | 10.25 |
| Genome assembly | Haplotype 1 | Haplotype 2 |
|---|---|---|
| Assembly name | idLepBrev1.hap1.1 | idLepBrev1.hap2.1 |
| Assembly accession | GCA_964036015.1 | GCA_964036025.1 |
| Assembly level | chromosome | scaffold |
| Span (Mb) | 850.81 | 851.46 |
| Number of contigs | 467 | 437 |
| Number of scaffolds | 161 | 134 |
| Longest scaffold (Mb) | 232.65 | - |
| Assembly metrics (benchmark) | Haplotype 1 | Haplotype 2 |
| Contig N50 length (≥ 1 Mb) | 5.29 Mb | 4.71 Mb |
| Scaffold N50 length (= chromosome N50) | 155.12 Mb | 155.98 Mb |
| Consensus quality (QV) (≥ 40) | 61.6 | 61.8 |
|
| 89.03% | 89.15% |
| Combined
| 99.66% | |
| BUSCO* (S > 90%; D < 5%) | C:96.9%[S:95.4%,D:1.4%],F:0.8%,M:2.4%,n:3285 | - |
| Percentage of assembly assigned to
| 99.05% | - |
| Sex chromosomes (localised homologous
| Not identified | - |
| Organelles (one complete allele) | Mitochondrial genome: 18.16 kb | - |
| INSDC accession | Name | Length (Mb) | GC% |
|---|---|---|---|
| 1 | 232.65 | 37 | |
| 2 | 162.87 | 37 | |
| 3 | 155.12 | 37 | |
| 4 | 148.83 | 37.5 | |
| 5 | 117.46 | 37.5 | |
| 6 | 25.79 | 37.5 | |
| MT | 0.02 | 21 |
| Software tool | Version | Source |
|---|---|---|
| BLAST | 2.14.0 |
|
| BlobToolKit | 4.3.9 |
|
| BUSCO | 5.5.0 |
|
| bwa-mem2 | 2.2.1 |
|
| DIAMOND | 2.1.8 |
|
| fasta_windows | 0.2.4 |
|
| FastK | 666652151335353eef2fcd58880bcef5bc2928e1 |
|
| Gfastats | 1.3.6 |
|
| GoaT CLI | 0.2.5 |
|
| Hifiasm | 0.19.8-r603 |
|
| HiGlass | 44086069ee7d4d3f6f3f0012569789ec138f42b8
|
|
| MerquryFK | d00d98157618f4e8d1a9190026b19b471055b22e |
|
| Minimap2 | 2.24-r1122 |
|
| MitoHiFi | 3 |
|
| MultiQC | 1.14, 1.17, and 1.18 |
|
| Nextflow | 23.10.0 |
|
| PretextView | 0.2.5 |
|
| PretextSnapshot | - |
|
| samtools | 1.19.2 |
|
| sanger-tol/ascc | 0.1.0 |
|
| sanger-tol/
| 0.6.0 |
|
| Seqtk | 1.3 |
|
| Singularity | 3.9.0 |
|
| TreeVal | 1.2.0 |
|
| YaHS | 1.2a.2 |
|
- —Wellcome Trust
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDiptera species taxonomy and behavior · Viral Infections and Vectors · Species Distribution and Climate Change
Species taxonomy
Eukaryota; Opisthokonta; Metazoa; Eumetazoa; Bilateria; Protostomia; Ecdysozoa; Panarthropoda; Arthropoda; Mandibulata; Pancrustacea; Hexapoda; Insecta; Dicondylia; Pterygota; Neoptera; Endopterygota; Diptera; Brachycera; Muscomorpha; Asiloidea; Asilidae; Brachyrhopalinae; Leptarthrus; Leptarthrus brevirostris (Meigen, 1804) (NCBI:txid468750)
Background
Leptarthrus brevirostris is a robberfly belonging to the family Asilidae. It is, for a robberfly, a small to moderate-sized species, the adult males having remarkably elongated, slender and flattened hind metatarsi, the next three segments being minute, a diagnostic feature. The hind tarsi of the females show no such peculiarity ( Stubbs & Drake, 2014). Otherwise, it is a hump-backed, blackish fly with partly red-orange tibiae.
The larvae are unknown but presumed to develop as predators in soil. Little is known about the prey of the adults, although there is an observation of braconid parasitic wasps being taken ( Hobby, 1930; Stubbs & Drake, 2014) and a suggestion they take bugs (Hemiptera) ( Falk, 2025). They prefer to hunt from wooden fence posts and the leaves of small bushes ( van den Broek & Schulten, 2017).
The species occurs widely across Britain (but not Ireland), central Europe and southern Scandinavia, with occasional records further east as far as Russia, including the Balkans ( GBIF Secretatiat, 2023).
Within Britain it occurs in different habitats in different geographical areas. In central southern and south-eastern England it is strongly associated with dry calcareous grasslands, especially those on chalk. In contrast, in south-west England, north-west Wales and much of Scotland it typically occurs within acid grasslands and open woodlands, often on poorly drained soils, as well as on base-enriched flushes ( Falk, 2025; Stubbs & Drake, 2014, and personal observations). The reasons for these habitat differences are not known, but may relate to the use of different larval hosts or adult prey ( Stubbs & Drake, 2014). Research is much needed on larval and adult behaviour and ecology.
As part of the Darwin Tree of Life Project – which aims to generate high-quality reference genomes for all named eukaryotic species in Britain and Ireland to support research, conservation, and the sustainable use of biodiversity – we present a chromosomally complete genome sequence for Leptarthrus brevirostris. This genome was assembled using the Tree of Life pipeline from a specimen collected in Kings Wood, London, England, United Kingdom ( Figure 1).
Photograph of the Leptarthrus brevirostris (idLepBrev1) specimen used for genome sequencing.
Genome sequence report
Sequencing data
The genome of a specimen of Leptarthrus brevirostris ( Figure 1) was sequenced using Pacific Biosciences single-molecule HiFi long reads, generating 25.80 Gb (gigabases) from 3.04 million reads, which were used to assemble the genome. GenomeScope analysis estimated the haploid genome size at 824.97 Mb, with a heterozygosity of 0.67% and repeat content of 29.15%. These estimates guided expectations for the assembly. Based on the estimated genome size, the sequencing data provided approximately 31 coverage. Hi-C sequencing produced 154.29 Gb from 1,021.82 million reads, used to scaffold the assembly. RNA sequencing data were also generated and are available in public sequence repositories. Table 1 summarises the specimen and sequencing details.
Table 1.: Specimen and sequencing data for Leptarthrus brevirostris.
Assembly statistics
The genome was assembled into two haplotypes using Hi-C phasing. Haplotype 1 was curated to chromosome level, while haplotype 2 was assembled to scaffold level. The assembly was improved by manual curation, which corrected 118 misjoins or missing joins. These interventions decreased the scaffold count by 17.77%. The final assembly has a total length of 850.81 Mb in 161 scaffolds, with 306 gaps, and a scaffold N50 of 155.12 Mb ( Table 2).
Table 2.: Genome assembly data for Leptarthrus brevirostris.
The snail plot in Figure 2 provides a summary of the assembly statistics, indicating the distribution of scaffold lengths and other assembly metrics. Figure 3 shows the distribution of scaffolds by GC proportion and coverage. Figure 4 presents a cumulative assembly plot, with separate curves representing different scaffold subsets assigned to various phyla, illustrating the completeness of the assembly.
Genome assembly of Leptarthrus brevirostris, idLepBrev1.hap1.1: metrics.The BlobToolKit snail plot provides an overview of assembly metrics and BUSCO gene completeness. The circumference represents the length of the whole genome sequence, and the main plot is divided into 1,000 bins around the circumference. The outermost blue tracks display the distribution of GC, AT, and N percentages across the bins. Scaffolds are arranged clockwise from longest to shortest and are depicted in dark grey. The longest scaffold is indicated by the red arc, and the deeper orange and pale orange arcs represent the N50 and N90 lengths. A light grey spiral at the centre shows the cumulative scaffold count on a logarithmic scale. A summary of complete, fragmented, duplicated, and missing BUSCO genes in the diptera_odb10 set is presented at the top right. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/GCA_964036015.1/dataset/GCA_964036015.1/snail.
Genome assembly of Leptarthrus brevirostris, idLepBrev1.hap1.1: BlobToolKit GC-coverage plot.Blob plot showing sequence coverage (vertical axis) and GC content (horizontal axis). The circles represent scaffolds, with the size proportional to scaffold length and the colour representing phylum membership. The histograms along the axes display the total length of sequences distributed across different levels of coverage and GC content. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/GCA_964036015.1/dataset/GCA_964036015.1/blob.
Genome assembly of Leptarthrus brevirostris, idLepBrev1.hap1.1: BlobToolKit cumulative sequence plot.The grey line shows cumulative length for all scaffolds. Coloured lines show cumulative lengths of scaffolds assigned to each phylum using the buscogenes taxrule. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/GCA_964036015.1/dataset/GCA_964036015.1/cumulative.
Most of the assembly sequence (99.05%) was assigned to 6 chromosomal-level scaffolds. These chromosome-level scaffolds, confirmed by Hi-C data, are named according to size ( Figure 5; Table 3). During curation, we noted that read coverage suggests this is the homogametic sex, but we did not identify the sex chromosome(s) as sequence data from the heterogametic sex was not available and homology is unreliable for sex chromosome identification in Diptera due to frequent sex chromosome turnover ( Vicoso & Bachtrog, 2015).
Genome assembly of Leptarthrus brevirostris.Hi-C contact map of the idLepBrev1.hap1.1 assembly, generated using PretextSnapshot. Chromosomes are shown in order of size and labelled with chromosome numbers (top) and chromosome accession numbers (left).
Table 3.: Chromosomal pseudomolecules in the genome assembly of Leptarthrus brevirostris, idLepBrev1.
The mitochondrial genome was also assembled. This sequence is included as a contig in the multifasta file of the genome submission and as a standalone record.
Assembly quality metrics
The estimated Quality Value (QV) and k-mer completeness metrics, along with BUSCO completeness scores, were calculated for each haplotype and the combined assembly. The QV reflects the base-level accuracy of the assembly, while k-mer completeness indicates the proportion of expected k-mers identified in the assembly. BUSCO scores provide a measure of completeness based on benchmarking universal single-copy orthologues.
For haplotype 1, the estimated QV is 61.6, and for haplotype 2, 61.8. When the two haplotypes are combined, the assembly achieves an estimated QV of 61.7. The k-mer completeness is 89.03% for haplotype 1 and 89.15% for haplotype 2; and 99.66% for the combined haplotypes. BUSCO v.5.5.0 analysis using the diptera_odb10 reference set ( n = 3,285) identified 96.9% of the expected gene set (single = 95.4%, duplicated = 1.4%) for haplotype 1.
Table 2 provides assembly metric benchmarks adapted from Rhie et al. (2021) and the Earth BioGenome Project Report on Assembly Standards September 2024. The haplotype 1 assembly achieves the EBP reference standard of 6.C.Q61.
Methods
Sample acquisition and DNA barcoding
The specimen used for genome sequencing was an adult female Leptarthrus brevirostris (specimen ID NHMUK014537452, ToLID idLepBrev1), from Porth Saxon swamp, Mawnan, Helford River, Cornwall, England, United Kingdom on 2021-06-28 by sweep netting. The specimen was collected and identified by Robert Wolton (Dipterists Forum) and preserved by dry freezing (–80 °C).
The initial identification was verified by an additional DNA barcoding process according to the framework developed by Twyford et al. (2024). A small sample was dissected from the specimen and stored in ethanol, while the remaining parts were shipped on dry ice to the Wellcome Sanger Institute (WSI) ( Pereira et al., 2022). The tissue was lysed, the COI marker region was amplified by PCR, and amplicons were sequenced and compared to the BOLD database, confirming the species identification ( Crowley et al., 2023). Following whole genome sequence generation, the relevant DNA barcode region was also used alongside the initial barcoding data for sample tracking at the WSI ( Twyford et al., 2024). The standard operating procedures for Darwin Tree of Life barcoding have been deposited on protocols.io ( Beasley et al., 2023).
Metadata collection for samples adhered to the Darwin Tree of Life project standards described by Lawniczak et al. (2022).
Nucleic acid extraction
The workflow for high molecular weight (HMW) DNA extraction at the Wellcome Sanger Institute (WSI) Tree of Life Core Laboratory includes a sequence of procedures: sample preparation and homogenisation, DNA extraction, fragmentation and purification ( Howard et al., 2025). Detailed protocols are available on protocols.io ( Denton et al., 2023b). The idLepBrev1 sample was prepared for DNA extraction by weighing and dissecting it on dry ice ( Jay et al., 2023). Tissue from the thorax and abdomen was homogenised using a PowerMasher II tissue disruptor ( Denton et al., 2023a).
HMW DNA was extracted in the WSI Scientific Operations core using the Automated MagAttract v2 protocol ( Oatley et al., 2023). The DNA was sheared into an average fragment size of 12–20 kb in a Megaruptor 3 system ( Bates et al., 2023). Sheared DNA was purified by solid-phase reversible immobilisation, using AMPure PB beads to eliminate shorter fragments and concentrate the DNA ( Strickland et al., 2023). The concentration of the sheared and purified DNA was assessed using a Nanodrop spectrophotometer and Qubit Fluorometer using the Qubit dsDNA High Sensitivity Assay kit. Fragment size distribution was evaluated by running the sample on the FemtoPulse system.
RNA was extracted from thorax and abdomen tissue of idLepBrev1 in the Tree of Life Laboratory at the WSI using the RNA Extraction: Automated MagMax™ mirVana protocol ( do Amaral et al., 2023). The RNA concentration was assessed using a Nanodrop spectrophotometer and a Qubit Fluorometer using the Qubit RNA Broad-Range Assay kit. Analysis of the integrity of the RNA was done using the Agilent RNA 6000 Pico Kit and Eukaryotic Total RNA assay.
Hi-C sample preparation and crosslinking
Hi-C data were generated from the head of the idLepBrev1 sample using the Arima-HiC v2 kit (Arima Genomics) with 20–50 mg of frozen tissue (stored at –80 °C). As per manufacturer’s instructions, tissue was fixed, and the DNA crosslinked using a TC buffer with 22% formaldehyde concentration, and a final formaldehyde concentration of 2%. The tissue was then homogenised using the Diagnocine Power Masher-II. The crosslinked DNA was digested using a restriction enzyme master mix, then biotinylated and ligated. A clean up was performed with SPRIselect beads prior to library preparation. DNA concentration was quantified using the Qubit Fluorometer v4.0 (Thermo Fisher Scientific) and Qubit HS Assay Kit, and sample biotinylation percentage was estimated using the Arima-HiC v2 QC beads.
Library preparation and sequencing
Library preparation and sequencing were performed at the WSI Scientific Operations core.
** PacBio HiFi **
At a minimum, samples were required to have an average fragment size exceeding 8 kb and a total mass over 400 ng to proceed to the low-input SMRTbell Prep Kit 3.0 protocol (Pacific Biosciences), depending on genome size and sequencing depth required. Libraries were prepared using the SMRTbell Prep Kit 3.0 as per the manufacturer's instructions. The kit includes the reagents required for end repair/A-tailing, adapter ligation, post-ligation SMRTbell bead cleanup, and nuclease treatment. Size-selection and clean-up were carried out using diluted AMPure PB beads (Pacific Biosciences). DNA concentration was quantified using the Qubit Fluorometer v4.0 (ThermoFisher Scientific) with Qubit 1X dsDNA HS assay kit and the final library fragment size analysis was carried out using the Agilent Femto Pulse Automated Pulsed Field CE Instrument (Agilent Technologies) and the gDNA 55kb BAC analysis kit.
Samples were sequenced on a Revio instrument (Pacific Biosciences, California, USA). Prepared libraries were normalised to 2 nM, and 15 μL was used for making complexes. Primers were annealed and polymerases were bound to create circularised complexes according to manufacturer’s instructions. The complexes were purified with the 1.2X clean up with SMRTbell beads. The purified complexes were then diluted to the Revio loading concentration (in the range 200–300 pM), and spiked with a Revio sequencing internal control. Samples were sequenced on Revio 25M SMRT cells (Pacific Biosciences, California, USA). The SMRT link software, a PacBio web-based end-to-end workflow manager, was used to set-up and monitor the run, as well as perform primary and secondary analysis of the data upon completion.
** Hi-C **
For Hi-C library preparation, the biotinylated DNA constructs were fragmented using a Covaris E220 sonicator and size-selected to 400–600 bp using SPRISelect beads. DNA was then enriched using Arima-HiC v2 Enrichment beads. The NEBNext Ultra II DNA Library Prep Kit (New England Biolabs) was used for end repair, A-tailing, and adapter ligation, following a modified protocol in which library preparation is carried out while the DNA remains bound to the enrichment beads. PCR amplification was performed using KAPA HiFi HotStart mix and custom dual-indexed adapters (Integrated DNA Technologies) in a 96-well plate format. Depending on sample concentration and biotinylation percentage determined at the crosslinking stage, samples were amplified for 10–16 PCR cycles. Post-PCR clean-up was carried out using SPRISelect beads. The libraries were quantified using the Accuclear Ultra High Sensitivity dsDNA Standards Assay kit (Biotium) and normalised to 10 ng/μL before sequencing. Hi-C sequencing was performed on the Illumina NovaSeq 6000 instrument with 150 bp paired-end reads.
** RNA **
Poly(A) RNA-Seq libraries were constructed using the NEB Ultra II RNA Library Prep kit, following the manufacturer’s instructions. RNA sequencing was performed on the Illumina NovaSeq 6000 instrument.
Genome assembly, curation and evaluation
** Assembly **
Prior to assembly of the PacBio HiFi reads, a database of k-mer counts ( k = 31) was generated from the filtered reads using FastK. GenomeScope2 ( Ranallo-Benavidez et al., 2020) was used to analyse the k-mer frequency distributions, providing estimates of genome size, heterozygosity, and repeat content.
The HiFi reads were assembled using Hifiasm in Hi-C phasing mode ( Cheng et al., 2021; Cheng et al., 2022), resulting in a pair of haplotype-resolved assemblies. The Hi-C reads ( Rao et al., 2014) were mapped to the primary contigs using bwa-mem2 ( Vasimuddin et al., 2019). The contigs were further scaffolded with Hi-C data in YaHS ( Zhou et al., 2023), using the --break option for handling potential misassemblies. The scaffolded assemblies were evaluated using Gfastats ( Formenti et al., 2022), BUSCO ( Manni et al., 2021) and MERQURY.FK ( Rhie et al., 2020).
The mitochondrial genome was assembled using MitoHiFi ( Uliano-Silva et al., 2023), which runs MitoFinder ( Allio et al., 2020) and uses these annotations to select the final mitochondrial contig and to ensure the general quality of the sequence.
** Assembly curation **
The assembly was decontaminated using the Assembly Screen for Cobionts and Contaminants (ASCC) pipeline. Flat files and maps used in curation were generated via the TreeVal pipeline ( Pointon et al., 2023). Manual curation was conducted primarily in PretextView ( Harry, 2022) and HiGlass ( Kerpedjiev et al., 2018), with additional insights provided by JBrowse2 ( Diesh et al., 2023). Scaffolds were visually inspected and corrected as described by Howe et al. (2021). Any identified contamination, missed joins, and mis-joins were amended, and duplicate sequences were tagged and removed. The curation process is documented at https://gitlab.com/wtsi-grit/rapid-curation.
** Assembly quality assessment **
The Merqury.FK tool ( Rhie et al., 2020), run in a Singularity container ( Kurtzer et al., 2017), was used to evaluate k-mer completeness and assembly quality for both haplotypes using the k-mer databases ( k = 31) computed prior to genome assembly. The analysis outputs included assembly QV scores and completeness statistics.
The genome was analysed in the blobtoolkit pipeline, a Nextflow ( Di Tommaso et al., 2017) port of the previous Snakemake Blobtoolkit pipeline ( Challis et al., 2020). It aligns the PacBio reads in SAMtools ( Danecek et al., 2021) and minimap2 ( Li, 2018) and generates coverage tracks for regions of fixed size. In parallel, it queries the GoaT database ( Challis et al., 2023) to identify all matching BUSCO lineages to run BUSCO ( Manni et al., 2021). For the three domain-level BUSCO lineages, the pipeline aligns the BUSCO genes to the UniProt Reference Proteomes database ( Bateman et al., 2023) with DIAMOND blastp ( Buchfink et al., 2021). The genome is also divided into chunks according to the density of the BUSCO genes from the closest taxonomic lineage, and each chunk is aligned to the UniProt Reference Proteomes database using DIAMOND blastx. Genome sequences without a hit are chunked using seqtk and aligned to the NT database with blastn ( Altschul et al., 1990). The blobtools suite combines all these outputs into a blobdir for visualisation.
The blobtoolkit pipeline was developed using nf-core tooling ( Ewels et al., 2020) and MultiQC ( Ewels et al., 2016), relying on the Conda package manager, the Bioconda initiative ( Grüning et al., 2018), the Biocontainers infrastructure ( da Veiga Leprevost et al., 2017), as well as the Docker ( Merkel, 2014) and Singularity ( Kurtzer et al., 2017) containerisation solutions.
Table 4 contains a list of relevant software tool versions and sources.
Wellcome Sanger Institute – Legal and Governance
The materials that have contributed to this genome note have been supplied by a Darwin Tree of Life Partner. The submission of materials by a Darwin Tree of Life Partner is subject to the ‘Darwin Tree of Life Project Sampling Code of Practice’, which can be found in full on the Darwin Tree of Life website here. By agreeing with and signing up to the Sampling Code of Practice, the Darwin Tree of Life Partner agrees they will meet the legal and ethical requirements and standards set out within this document in respect of all samples acquired for, and supplied to, the Darwin Tree of Life Project.
Further, the Wellcome Sanger Institute employs a process whereby due diligence is carried out proportionate to the nature of the materials themselves, and the circumstances under which they have been/are to be collected and provided for use. The purpose of this is to address and mitigate any potential legal and/or ethical implications of receipt and use of the materials as part of the research project, and to ensure that in doing so we align with best practice wherever possible. The overarching areas of consideration are:
• Ethical review of provenance and sourcing of the material
• Legality of collection, transfer and use (national and international)
Each transfer of samples is further undertaken according to a Research Collaboration Agreement or Material Transfer Agreement entered into by the Darwin Tree of Life Partner, Genome Research Limited (operating as the Wellcome Sanger Institute), and in some circumstances other Darwin Tree of Life collaborators.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Allio R Schomaker-Bastos A Romiguier J : Mito Finder: efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol Ecol Resour. 2020;20(4):892–905. 10.1111/1755-0998.13160 32243090 PMC 7497042 · doi ↗ · pubmed ↗
- 2Altschul SF Gish W Miller W : Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. 10.1016/S 0022-2836(05)80360-2 2231712 · doi ↗ · pubmed ↗
- 3Bateman A Martin MJ Orchard S : Uni Prot: the universal protein knowledgebase in 2023. Nucleic Acids Res. 2023;51(D 1):D 523–D 531. 10.1093/nar/gkac 1052 36408920 PMC 9825514 · doi ↗ · pubmed ↗
- 4Bates A Clayton-Lucey I Howard C : Sanger Tree of Life HMW DNA fragmentation: diagenode Megaruptor ®3 for LI Pac Bio. protocols.io. 2023. 10.17504/protocols.io.81wgbxzq 3lpk/v 1 · doi ↗
- 5Beasley J Uhl R Forrest LL : DNA barcoding SO Ps for the Darwin Tree of Life project. protocols.io. 2023; [Accessed 25 June 2024]. 10.17504/protocols.io.261ged 91jv 47/v 1 · doi ↗
- 6Buchfink B Reuter K Drost HG : Sensitive protein alignments at Tree-of-Life scale using DIAMOND. Nat Methods. 2021;18(4):366–368. 10.1038/s 41592-021-01101-x 33828273 PMC 8026399 · doi ↗ · pubmed ↗
- 7Challis R Kumar S Sotero-Caio C : Genomes on a Tree (Goa T): a versatile, scalable search engine for genomic and sequencing project metadata across the eukaryotic Tree of Life [version 1; peer review: 2 approved]. Wellcome Open Res. 2023;8:24. 10.12688/wellcomeopenres.18658.1 36864925 PMC 9971660 · doi ↗ · pubmed ↗
- 8Challis R Richards E Rajan J : Blob Tool Kit – interactive quality assessment of genome assemblies. G 3 (Bethesda). 2020;10(4):1361–1374. 10.1534/g 3.119.400908 32071071 PMC 7144090 · doi ↗ · pubmed ↗