A Mild Juvenile Onset Canavan Disease With Atypical Clinical Presentation and MRI Brain Features
Preeya Rehsi, Ata Siddiqui, Rahul Singh, Obioma Ihezue, Rebecca Halligan

TL;DR
A 13-year-old girl showed mild, unusual symptoms of Canavan disease, highlighting the need to recognize atypical cases for accurate diagnosis.
Contribution
This case expands the known clinical and radiological spectrum of Canavan disease with a mild and atypical presentation.
Findings
The patient exhibited intention tremor and fine motor difficulties instead of typical CD symptoms.
MRI showed symmetrical brain changes initially suggesting a different disorder.
Whole genome sequencing confirmed the diagnosis of Canavan disease.
Abstract
This case report highlights an atypical presentation of Canavan disease (CD) in a 13‐year‐old female with intention tremor and fine motor difficulties. Neuroimaging revealed symmetrical changes in various brain regions initially suggesting a neurometabolic or mitochondrial disorder. However, further investigations, including biochemical analysis and whole genome sequencing, confirmed a diagnosis of CD. Unlike classical presentations, this case exhibited milder symptoms and unusual MRI findings, contributing to the expanding clinical and radiological spectrum of CD. The importance of recognizing such atypical presentations is emphasized for accurate diagnosis and management of CD.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
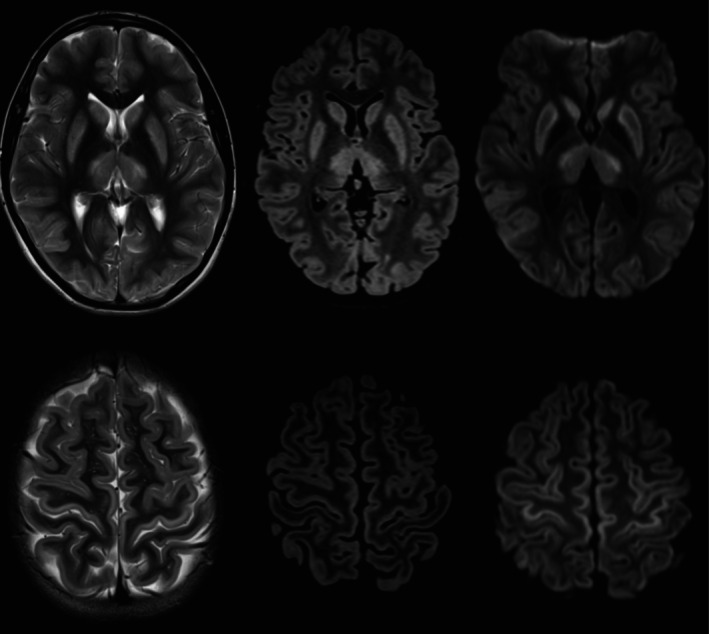 FIGURE 1
FIGURE 1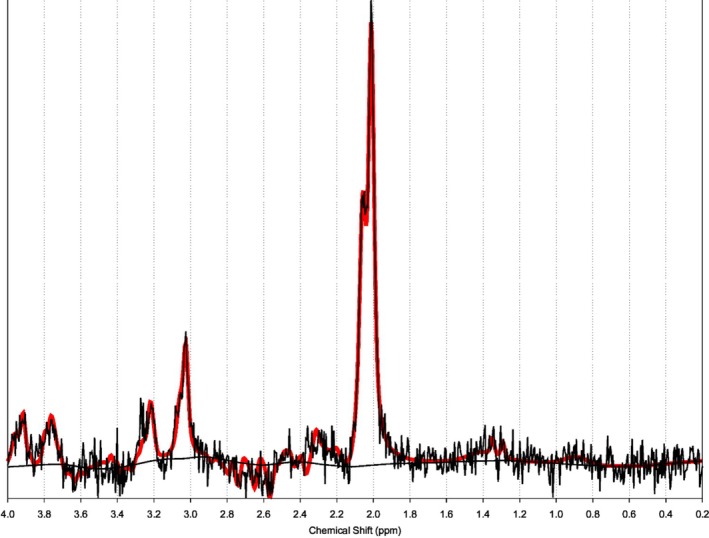 FIGURE 2
FIGURE 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced MRI Techniques and Applications · Atomic and Subatomic Physics Research · Fetal and Pediatric Neurological Disorders
Summary
- Milder cases of Canavan disease can present with atypical neuroimaging features, broadening the clinical and radiological phenotype.
Case Report
1
A well 13‐year‐old female presented with a 5‐year history of intention tremor and difficulty with fine motor skills. MRI brain revealed symmetrical changes in the putamen, thalami, caudate nuclei and occipital cortex bilaterally, raising the possibility a neurometabolic or mitochondrial disorder (Figure 1). Biochemical investigations revealed a normal lactate, acylcarnitine profile and plasma amino acids. Urine organic acid analysis revealed mildly elevated N‐acetyl aspartic acid (NAA). Trio whole genome sequencing confirmed homozygosity for the ASPA c.820G>A p.(Gly274Arg) likely pathogenic variant, consistent with a diagnosis of Canavan disease (CD).
MRI with axial T2W, FLAIR and DWI images taken at the level of the basal ganglia (top row) and towards the vertex (bottom row) showing bilateral symmetrical basal ganglia, thalamic and cortical signal changes. The cortical changes are more evident within the precentral gyri (primary motor cortex). Subtle early subcortical white matter involvement is seen in the occipital lobes.
After 12 months of follow up there is no symptom progression. However, she has developed new onset Type 1 Diabetes Mellitus with classical symptoms of polyuria, polydipsia, weight loss, and mild diabetic ketoacidosis at presentation. An interval MRI brain with spectroscopy showed stable appearances with an elevated NAA peak on MRS (Figure 2). In hindsight, there was early subcortical white matter involvement in the occipital lobes.
Single voxel MR spectroscopy at TE 288 ms taken from the basal ganglia shows a significantly elevated NAA peak.
Discussion
2
CD is classically a life‐limiting, neurodegenerative condition characterized by infantile onset severe neurodevelopmental delay followed by arrest, seizures, visual impairment, and death in early infancy [1]. There is, however, emerging evidence of clinical heterogeneity associated with ASPA variants [2].
This G274R variant found in our patient was first described by Shaag et al. in 1995 in the homozygous state, but presenting with a severe phenotype including axial hypotonia from 3 months of age with white matter changes on MRI brain [3]. The same variant was found in a Pakistani family with moderate to severe presentation of CD however their neuroimaging has not been described [4].
Subsequent reports of milder phenotypes related to the G274R variant have been described. A child of Greek origin developed seizures at 11 weeks of life but continued to show developmental progress and by 4 years of age is walking with support and speaking single words. T2 weighted imaging showed increased signals in the periphery of the thalami, putamina, head of the caudate nucleus, and subcortical white matter, as well as the tegmentum of the pons and the white matter around the 4th ventricle [2]. This patient had two separate homozygous variants, one being the G274R and the other K213E. The latter variant has been proven to express wildtype protein activity. Our case is comparable to a patient reported in Turkey in 2020 with a similar clinical symptom of an intention tremor presenting at 13 years of age. MRI brain showed symmetric hyperintensities isolated to the pons and this patient was homozygous for the G274R variant [5].
Classically on MRI, CD affects the white matter in a diffuse and symmetrical pattern, with bilateral involvement of the globus pallidus and less marked involvement of the cerebellum, thalami, and brainstem. There is diffusion restriction without contrast enhancement in the affected white matter [6]. A spongiform degeneration is classically described, separating CD from other leukodystrophies. The imaging findings in our patient are atypical, and our case contributes to the broadening clinical and radiological phenotype in CD, demonstrating the emergence of milder cases.
The interindividual variability seen among patients with the G274R variant and other variants may be attributable to unidentified epigenetic and environmental factors, and further research is required to explore this.
Author Contributions
As first author P.R. contributed to the design, acquisition of data, and drafting of the work. Co‐authors A.S. and R.S. provided interpretation and expertise in the neuroimaging aspects of the work. Co‐author O.I. participated in providing clinical detail. Senior author and guarantor R.H. participated in the conception, design, acquisition of data, critical review and revision of the work. All authors reviewed the work and provided final approval of the version to be published.
Ethics Statement
The authors have nothing to report.
Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from the patient for being included in this report.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1A. Nagy , A. E. Bley , and F. Eichler , “Canavan Disease,” in Gene Reviews((R)), ed. M. P. Adam , J. Feldman , G. M. Mirzaa , et al. (University of Washington, Seattle, 1993).20301412 · pubmed ↗
- 2U. Tacke , H. Olbrich , J. O. Sass , et al., “Possible Genotype‐Phenotype Correlations in Children With Mild Clinical Course of Canavan Disease,” Neuropediatrics 36, no. 4 (2005): 252–255.16138249 10.1055/s-2005-865865 · doi ↗ · pubmed ↗
- 3A. Shaag , Y. Anikster , E. Christensen , et al., “The Molecular Basis of Canavan (Aspartoacylase Deficiency) Disease in European Non‐Jewish Patients,” American Journal of Human Genetics 57, no. 3 (1995): 572–580.7668285 PMC 1801272 · pubmed ↗
- 4R. Hussain , S. Daud , N. Kakar , et al., “A Missense Mutation (p.G 274R) in Gene ASPA Causes Canavan Disease in a Pakistani Family,” Molecular Biology Reports 39, no. 5 (2012): 6197–6201, 10.1007/s 11033-011-1438-2.22219087 · doi ↗ · pubmed ↗
- 5N. E. Çakar and T. Aksu Uzunhan , “A Case of Juvenile Canavan Disease with Distinct Pons Involvement,” Brain and Development 42, no. 2 (2020): 222–225, 10.1016/j.braindev.2019.11.009.31839386 · doi ↗ · pubmed ↗
- 6M. S. Knaap and J. Valk , Magnetic Resonance of Myelin, Myelination, and Myelin Disorders (Springer, 1995).