Malignant paraganglioma of the kidney: a rare surgical case with 3-year disease-free survival
Fu-Xiang Lin, Pengpeng Zhao, Le Xie, Zhan-Ping Xu

TL;DR
A 36-year-old woman with a rare kidney tumor underwent surgery and remained cancer-free for three years without additional treatment.
Contribution
This case highlights successful surgical management of a rare malignant kidney paraganglioma without adjuvant therapy.
Findings
A 14 cm renal mass was surgically removed and confirmed as a malignant paraganglioma.
No recurrence was observed after 3 years of follow-up with PET-CT.
The case suggests surgery alone may be sufficient for select localized malignant paragangliomas.
Abstract
A 36-year-old woman presented with abdominal pain and a left renal mass on imaging. Laboratory findings showed anemia (Hb 88 g/l), leukocytosis (12.31 × 109/l), and pyuria. Computed tomography (CT) revealed a 14 cm left renal cystic-solid mass suspicious for malignancy. Radical nephrectomy was performed, retrieving a 2600 g tumor. Histopathology demonstrated malignant paraganglioma with expansive growth confined to renal parenchyma. No adjuvant therapy was administered. At 3-year follow-up, surveillance positron emission tomography-computed tomography (PET-CT) confirmed no recurrence or metastasis. Renal paragangliomas represent <1% of such tumors, with malignant variants posing diagnostic and therapeutic challenges. This case underscores surgical resection as definitive management for localized disease and suggests favorable outcomes are achievable without adjuvant treatment in select…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
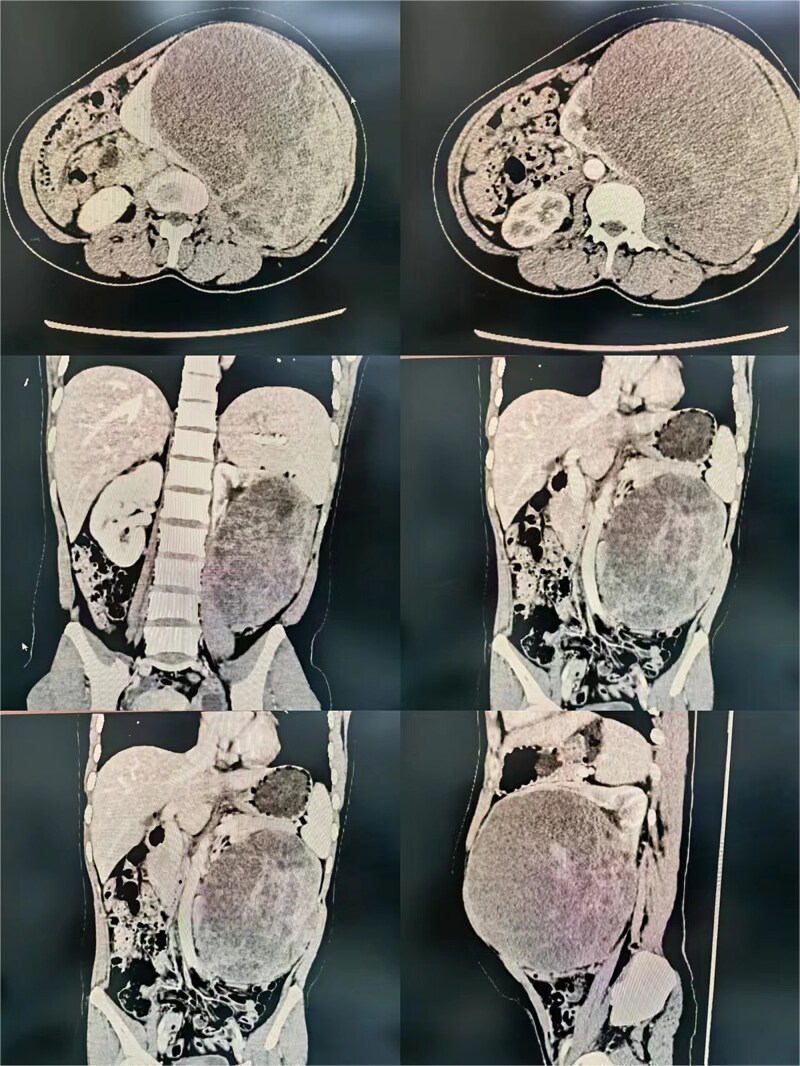 Figure 1
Figure 1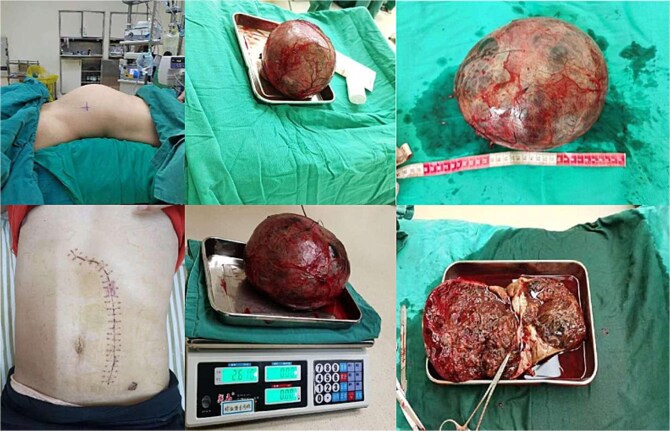 Figure 2
Figure 2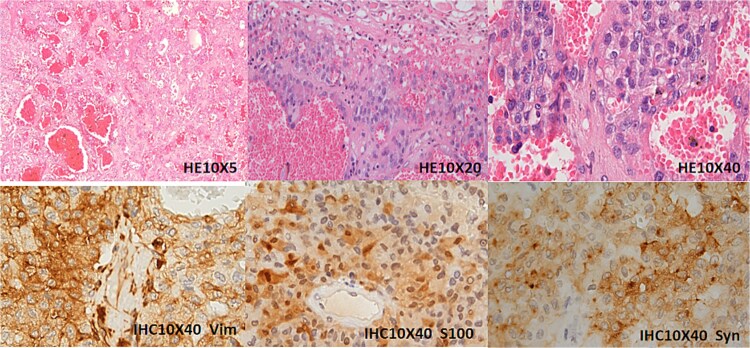 Figure 3
Figure 3- —2023 Medical Research Project of Foshan Health Bureau
- —2025 Annual Guangdong Provincial Administration of Traditional Chinese Medicine Scientific Research Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdrenal and Paraganglionic Tumors · Renal cell carcinoma treatment · Hormonal Regulation and Hypertension
Introduction
Paragangliomas are neuroendocrine tumors arising from autonomic nervous system ganglia, with >85% occurring in the adrenal medulla. Primary renal involvement is exceptionally rare, accounting for <1% of cases [1]. Malignant transformation occurs in <10% overall, but remains poorly characterized at renal sites due to scant literature [2]. We detail a surgically managed malignant renal paraganglioma with sustained remission at 3 years, examining implications for management strategies.
Case report
A 36-year-old female with no comorbidities presented with progressive abdominal pain for 1 month. Initial imaging suggested pancreatic origin, but contrast-enhanced CT localized a 14 × 13 × 12 cm heterogeneous left renal mass concerning for malignancy (Fig. 1). Laboratory workup revealed anemia (Hb 88 g/l), leukocytosis (12.31 × 10^9^/l), and sterile pyuria (WBC 814.1/μl). No hormonal hypersecretion was documented.
Radiological imaging of the left renal mass.
Surgical procedure
Following right lateral decubitus positioning, a transperitoneal approach via left paramedian incision exposed a retroperitoneal mass displacing bowel superiority. Intraoperative findings confirmed renal parenchymal origin without adjacent organ
involvement. The renal artery and vein were sequentially ligated using surgical clips and silk sutures. Complete resection required en bloc nephrectomy with ureteral division. Operative time was 180 minutes with 500 ml blood loss necessitating transfusion (Fig. 2).
Gross image of the resected left renal mass.
Pathologic findings
Gross examination revealed a 2600 g encapsulated mass replacing 80% of renal parenchyma (Fig. 2). Histopathology demonstrated nested neoplastic cells with eosinophilic cytoplasm, nuclear atypia, and vascular invasion. Immunohistochemistry confirmed synaptophysin (+), chromogranin A (+), and Ki-67 index 15% (Fig. 3). These features supported malignant paraganglioma (World Health Organization Grade 1) with negative resection margins.
Immunohistochemical analysis of the left renal mass. The tumor is situated within the renal parenchyma, compressing and displacing adjacent renal tubules with associated atrophy. Histologically, it demonstrates nested and papillary growth patterns within a richly vascularized stroma. High-power examination reveals tumor cells with vacuolated or eosinophilic cytoplasm, prominent small nucleoli, and rare mitotic figures. Immunohistochemical staining shows diffuse cytoplasmic positivity for Vimentin (Vim), S100, and Synaptophysin (Syn).
Discussion
This report highlights three critical aspects of renal paraganglioma management.
Diagnostic considerations
Renal paragangliomas frequently mimic renal cell carcinoma or retroperitoneal sarcomas radiographically. In our case, initial misattribution to pancreatic origin underscores diagnostic pitfalls. Preoperative biopsy is discouraged due to bleeding risk and sampling limitations [3]. Elevated leukocyte count observed herein may reflect tumor necrosis, previously unreported in renal cases.
Surgical strategy
Radical nephrectomy remains the cornerstone treatment [4]. The transperitoneal approach facilitated safe dissection despite tumor bulk, avoiding intraoperative hemodynamic instability seen in functional pheochromocytomas. Unlike adrenal paragangliomas requiring adrenal vein-first ligation, renal venous control followed arterial occlusion without complications.
Long-term outcomes
Our patient’s 3-year disease-free status contrasts with reported 40%–50% metastatic rates in malignant cases [5]. This discrepancy may reflect complete excision before vascular spread. The 15% Ki-67 index fell below the 20% cutoff predicting aggressive behavior in extra-adrenal paragangliomas [5], supporting surveillance-only post-resection. Serial PET-CT remains essential given late recurrence risks.
Conclusion
Malignant renal paraganglioma, though rare, should be considered in large hypervascular renal masses. Open nephrectomy provides definitive therapy with potential for extended recurrence-free survival even in malignant variants. Avoiding adjuvant treatment in cases with favorable histological features warrants a prospective study.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Neumann HPH, Young WF, Eng C. Pheochromocytoma and paraganglioma. New Engl J Med 2019;381:552–65. 10.1056/NEJ Mra 180665131390501 · doi ↗ · pubmed ↗
- 2Jha A, Taïeb D, Carrasquillo JA, et al. High-specific-activity-131I-MIBG versus 177Lu-DOTATATE targeted radionuclide therapy for metastatic pheochromocytoma and paraganglioma. Clin Cancer Res 2021;27:2989–95. 10.1158/1078-0432.CCR-20-370333685867 PMC 8172462 · doi ↗ · pubmed ↗
- 3Trans-Atlantic RPS Working Group . Management of primary retroperitoneal sarcoma (RPS) in the adult: a consensus approach from the trans-Atlantic RPS working group. Ann Surg Oncol 2015;22:256–63. 10.1245/s 10434-014-3965-225316486 · doi ↗ · pubmed ↗
- 4Lenders JWM, Kerstens MN, Amar L, et al. Management and future directions of research of phaeochromocytoma and paraganglioma: a position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J Hypertens 2020;38:1443–56. 10.1097/HJH.000000000000243832412940 PMC 7486815 · doi ↗ · pubmed ↗
- 5Jimenez C, Ma J, Roman Gonzalez A, et al. TNM staging and overall survival in patients with pheochromocytoma and sympathetic paraganglioma. J Clin Endocr Metab 2023;108:1132–42. 10.1210/clinem/dgac 67736433823 · doi ↗ · pubmed ↗